製造業者は機器のMDRの適合を行う場合、適合性評価手順(Conformity Assessment Procedures)に基づく必要がある。
製造業者は、医療機器のリスクに応じた「適合性評価手順」を見極める必要がある。
適合性評価手順はArticle 52 「適合性評価手順」に記載されている。
「製造業者は、機器を上市する前に、当該機器の適合性評価を、Annex Ⅸ~Ⅺ に規定されている該当する適合性評価手順に従って実施しなければならない。」
適合性評価手順は機器のクラス等により異なる。
- 日本、米国では、行政当局(または権限を委託された外部機関)が新しい医療機器について市販前の承認(認証)を与えるというシステムを採用 l製造業者の観点から見ると、医療機器の市販前に審査を受け、その結果として「承認(認証)を与えられる」形となる(受動形)
- 欧州では製造業者が自ら規制要求事項への適合を「宣言」し、CEマーキングを表示するという考え方を採用(能動形)
機器のクラス分類がClass Is,Ir,ImまたはClass IIa以上の場合、製造業者が適合宣言をするためのプロセスの中でNBの適合性評価が必要
そのため、製造業者が完全に自らの意思のみで宣言できるわけではない。
しかしながら、最後は自ら宣言をして、製造業者の法的責任の下でCEマーキングを表示するのが欧州の考え方である。
そのため、CEマーキングは「取得する」という表現は使用しない。 「添付する」または「表示する」を使用する。
MDDと異なった適合性評価手順は以下のとおりである。
- Class IIb埋め込み医療機器
- クラスⅢとほぼ同じ扱いとなる。
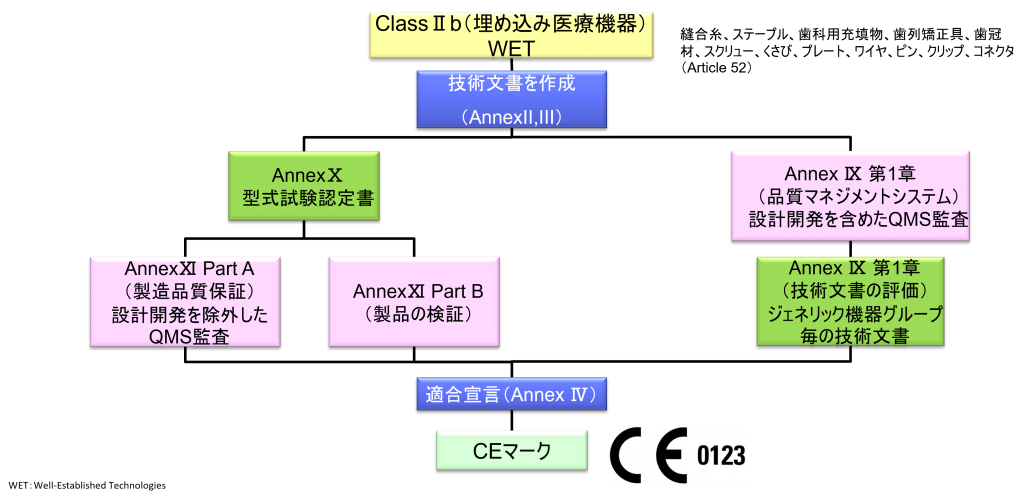
- ただし、縫合糸、ステープル、歯科用充填物、歯列矯正具、歯冠材、スクリュー、くさび、プレート、ワイヤ、ピン、クリップ、コネクタは除く
- ClassⅠ再使用可能な外科用器具
- クラスⅠ滅菌医療機器、測定機能を持つ医療機器とほぼ同じ扱いとなる
機器により、以下の特別な追加適合性手順がある。
- 医薬品を含む医療機器の場合、Annex Ⅸ Section 5.2またはAnnex X Section 6も適用すること。
所轄官庁(CA)または欧州医薬品庁(EMA)の評価がある。 - ヒト血液由来を含む機器は、出荷前にバッチ検証が必要。
- ヒト組織または細胞由来を含む医療機器の場合、Annex Ⅸ Section 5.3またはAnnex X Section 6も適用すること。
所轄官庁(CA)の評価がある。 - 人体開口部を経てまたは皮膚に適用され、人体に入れられ、人体内で吸収または局所的に分散されることを意図した物質、または物質のコンビネーション医療機器は、Annex Ⅸ Section 5.4 またはAnnex X Section 6も適用すること。
所轄官庁(CA)または欧州医薬品庁(EMA)の評価がある。
適合性評価手順(Article 52)
- 製造業者は、機器を上市する前に、当該機器の適合性評価を、Annex Ⅸ~Ⅺに規定されている該当する適合性評価手順に従って実施しなければならない。
- 製造業者は、上市されていない機器の使用を開始する前に、当該機器の適合性評価を、Annex Ⅸ~Ⅺに規定されている該当する適合性評価手順に従って実施しなければならない。
- クラスⅢの機器(ただし、カスタムメイド機器または治験機器は除く)の製造業者は、Annex Ⅸに定められているように、適合性評価の対象とならなければならない。あるいは、当該製造業者は、Annex Xに定められている適合性評価とAnnex Ⅺに定められている適合性評価を併せて適用することを選択してもよい。
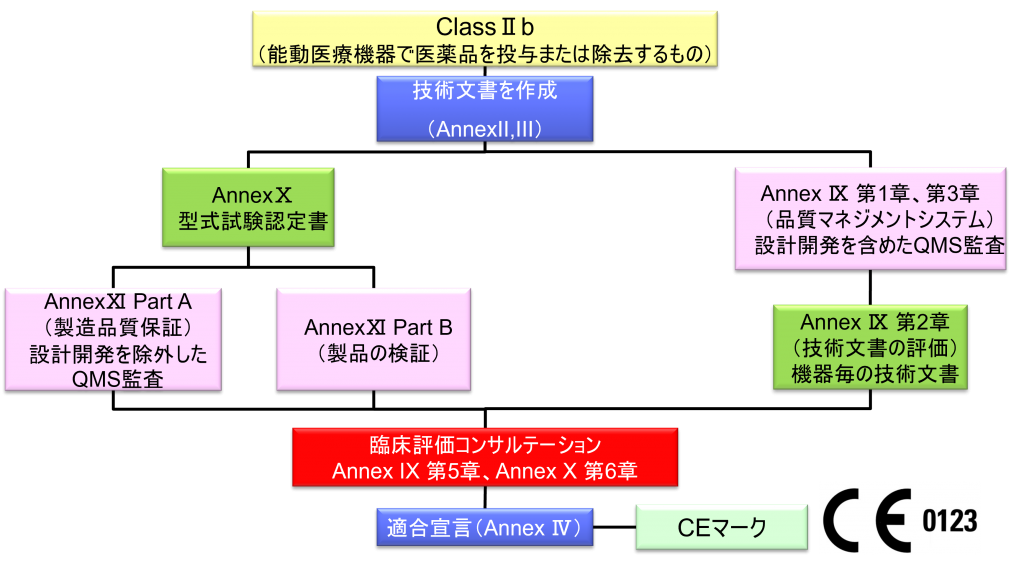
- クラスⅡb機器(ただし、カスタムメイド機器または治験機器は除く)の製造業者は、Annex Ⅸの第I章および第Ⅲ章に定められているように、適合性評価の対象とならなければならない。これには、ジェネリック機器グループごとに少なくとも一つの代表機器に関する、当該Annexの第4節に定められている技術文書の評価を含める。ただし、縫合糸、ステープル、歯科用充填剤、歯列矯正具、歯冠、ねじ、くさび、プレート、ワイヤ、ピン、クリップおよびコネクタを除くクラスⅡb埋込機器については、Annex Ⅸの第4節に定められているように、技術文書の評価は、全ての機器に適用しなければならない。あるいは、当該製造業者は、Annex Xに定められている型式審査に基づく適合性評価とAnnex Ⅺに定められている製品適合性検証に基づく適合性評価を併せて適用すること選択してもよい。
- 本条項の第4項の中段に挙げた免除機器に使用されている、確立した技術に類似した技術がその他のクラスⅡb埋込機器に使用される正当性が認められた場合、または患者、使用者などの健康と安全若しくはその他の公衆衛生面を保護する正当性が認められた場合、欧州委員会は、その他のクラスⅡb埋込機器のリストヘの追加、または機器のリストからの削除といった修正を行うために、Article 115に従って委任法令を採択する権限が与えられる。
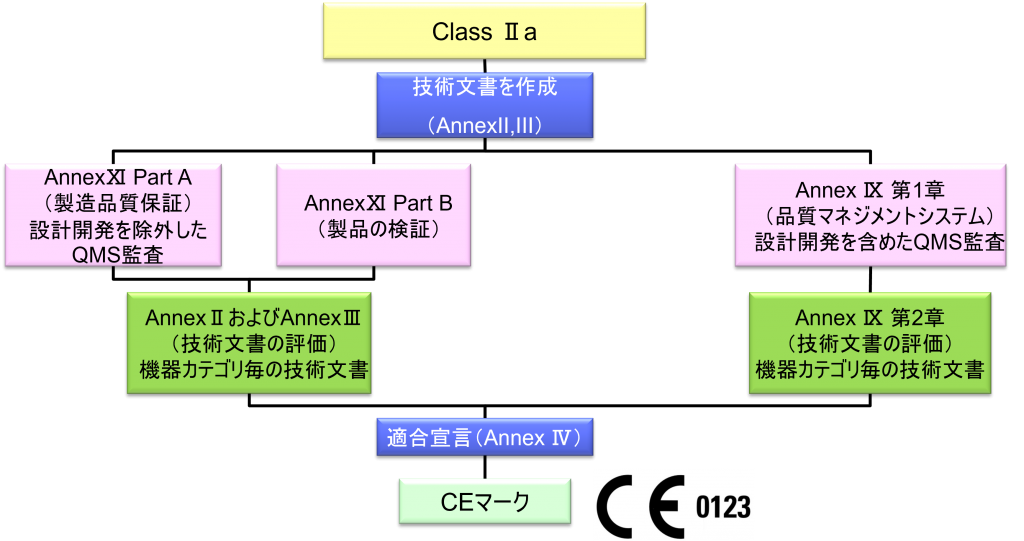
- クラスⅡa機器(ただし、カスタムメイド機器または治験機器は除く)の製造業者は、Annex Ⅸの第I章および第Ⅲ章に定められているように、適合性評価の対象とならなければならない。これには、機器のカテゴリーごとに少なくとも一つの代表機器に関する、当該Annex第4節に定められている技術文書の評価を含める。あるいは、当該製造業者は、Annex Ⅺの第10節または第18節に定められている適合性評価と併せて、AnnexⅡおよびⅢに規定されている技術文書を作成することを選択してもよい。技術文書の評価は、機器のカテゴリーごとに少なくとも一つの代表機器に対して適用しなければならない。
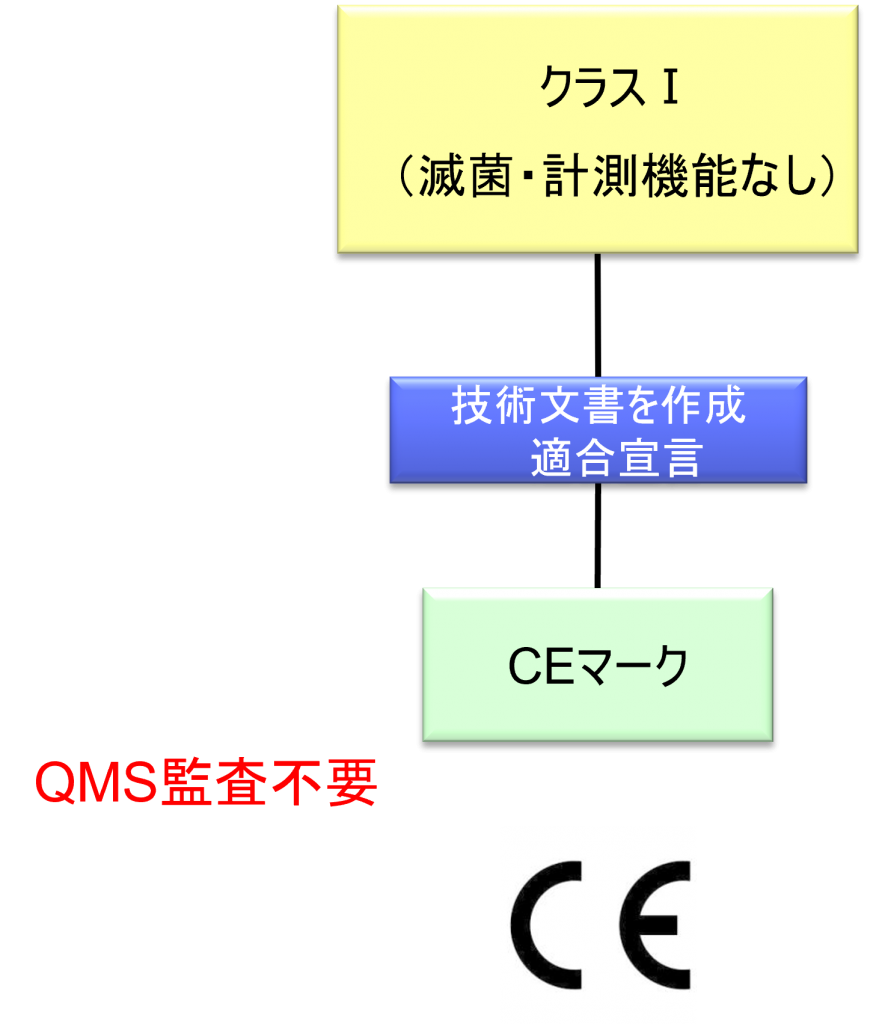
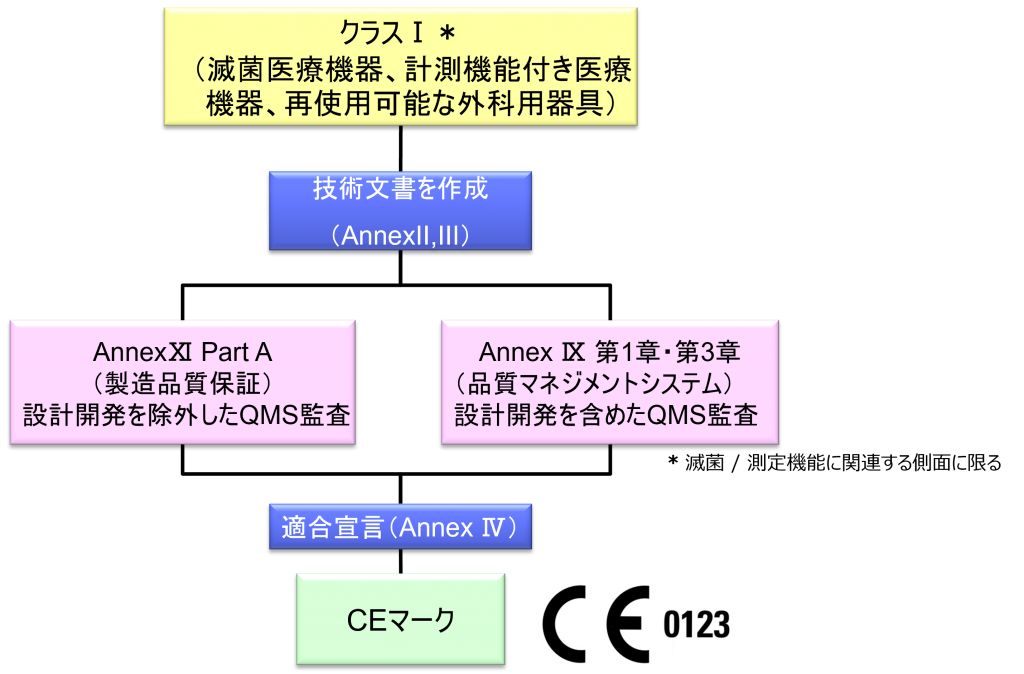
- クラスI機器(ただし、カスタムメイド機器または治験機器は除く)の製造業者は、AnnexⅡおよびⅢに規定されている技術文書の作成後、Article 19に規定されているEU適合宣言書を発行して製品の適合性を宣言しなければならない。それらの機器が滅菌状態で上市される、計測機能を有する、または再使用可能な外科用器具である場合、製造業者は、Annex Ⅸの第I章および第Ⅲ章またはAnnex ⅪのパートAに規定されている手順を適用しなければならない。ただし、次の事項については、本手順におけるNBの関与は制限されなければならない。
a.滅菌状態で上市される機器の場合、滅菌条件の設定、確保および維持に関連する事項
b.計測機能を有する機器の場合、計量要件への適合性に関する事項
c.再使用可能な外科用器具の場合、機器の再使用、特に洗浄、消毒、滅菌、保守および機能検査、並びに該当する取扱説明書に関する事項 - カスタムメイド機器の製造業者は、Annex XIIIに規定されている手順に従い、そうした機器を上市する前に、当該Annexの第1節に規定されているステートメントを作成しなければならない。前段に準拠して適用される手順に加え、クラスⅢカスタムメイド埋込機器の製造業者は、Annex Ⅸの第1章に定められている適合性評価手順の対象とならなければならない。あるいは、当該製造業者はAnnex ⅪのパートAに定められている適合性評価の適用を選択してもよい。
- Article 1第8項の前段に規定されている機器の場合、本条項の第3、4、6または7項に準拠して適用される手順に加え、適用対象となる場合は、Annex Ⅸの第5.2節またはAnnex Xの第6節に規定する手順も適用しなければならない。
- Article 1第6項の(f)または(g)およびArticle 1第10項の前段に従って、本規制の対象である機器の場合、本条項の第3、4、6または7項に準拠して適用される手順に加え、適用対象となる場合は、Annex Ⅸの第5.3節またはAnnex Xの第6節に規定する手順も適用しなければならない。
- 人体開口部を介して人体へ入れる、または皮膚に塗布することが意図され、人体に吸収される、または局所的に分散される物質若しくは物質の組み合わせから成る機器の場合、第3、4、6または7項に準拠して適用される手順に加え、適用対象となる場合は、Annex Ⅸの第5.4節またはAnnex Xの第6節に定められている手順も適用しなければならない。
- NBが設立されている加盟国は、第1項~第7項および第9項~第11項に規定されている手順に関する全ての文書または特定の文書(技術文書、監査、評価および検査の報告書を含む)が、加盟国が決定した欧州連合の公用語版で入手できるよう要求してもよい。そのような要求事項を定めない場合、それらの文書は、NBの同意する欧州連合の公用語で提供されなければならない。
- 治験機器は、Article 62~Article 81に規定されている要求事項の対象としなければならない。
- 欧州委員会は、NBによる適合性評価手順の統一的な適用を保証することを目的として、実施法令により次の点に関する詳細な取り決めおよび手順を規定してもよい。
a.クラスⅡaおよびクラスⅡbの機器については、Annex Ⅸの第2.3節第3項および第3.5 節に、クラスⅡaの機器については、Annex Ⅺの第10.2節に規定されている代表機器の技術文書の評価の頻度およびサンプリング
b.機器のリスクのクラスおよび種類を考慮し、Annex Ⅸの第3.4節に従ってNBが実施する予告なしの現地監査および抜取試験の最低頻度
c.Annex Ⅸの第3.4節および第4.3節、Annex Xの第3節、並びにAnnex Ⅺの第15節に従った抜取試験、技術文書の評価および型式審査においてNBが実施する物理的試験、臨床試験またはその他の試験
前段に規定された当該実施法令は、Article 114第3項に規定されている審査手順に従って採択されなければならない。
クラスⅠ機器(滅菌・計測機能なし)の適合性評価ルート
クラスI*(滅菌医療機器、計測機能付き医療機器、再使用可能な外科用器具)機器の適合性評価ルート
Class Ⅱa
ClassⅡb (能動医療機器で医薬品を投与または除去するもの)
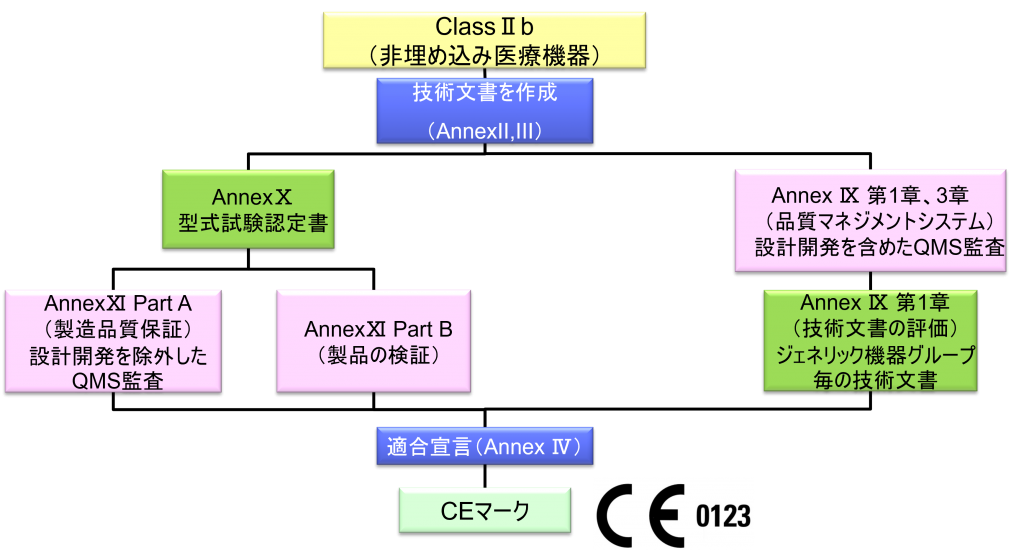
ClassⅡb(非埋め込み医療機器)
ClassⅡb(埋め込み医療機器)WET(Well-established Technology)
ClassⅡb(埋め込み医療機器)non-WET

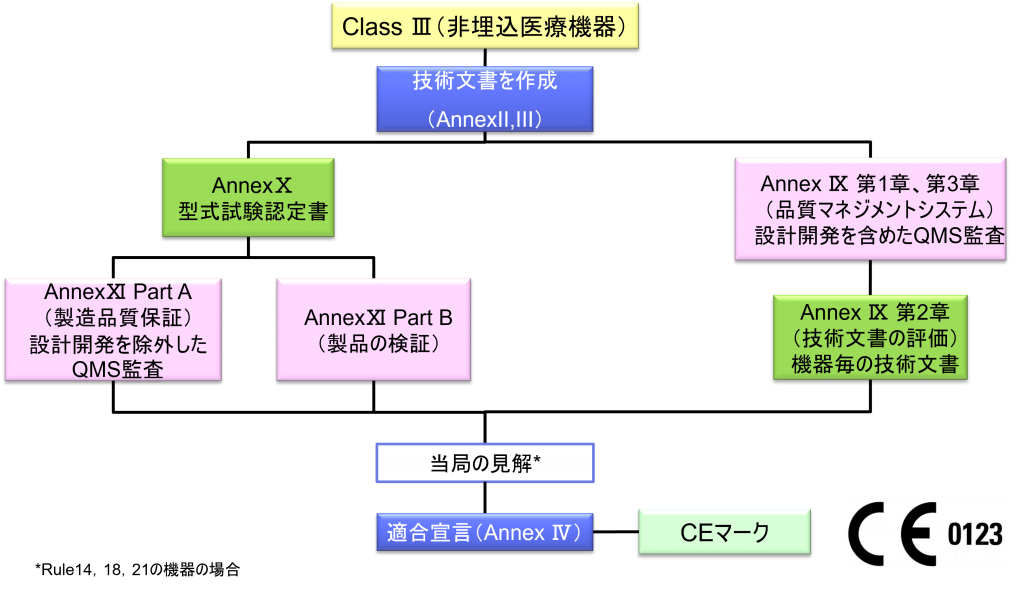
Class Ⅲ非埋込医療機器の適合性評価ルート
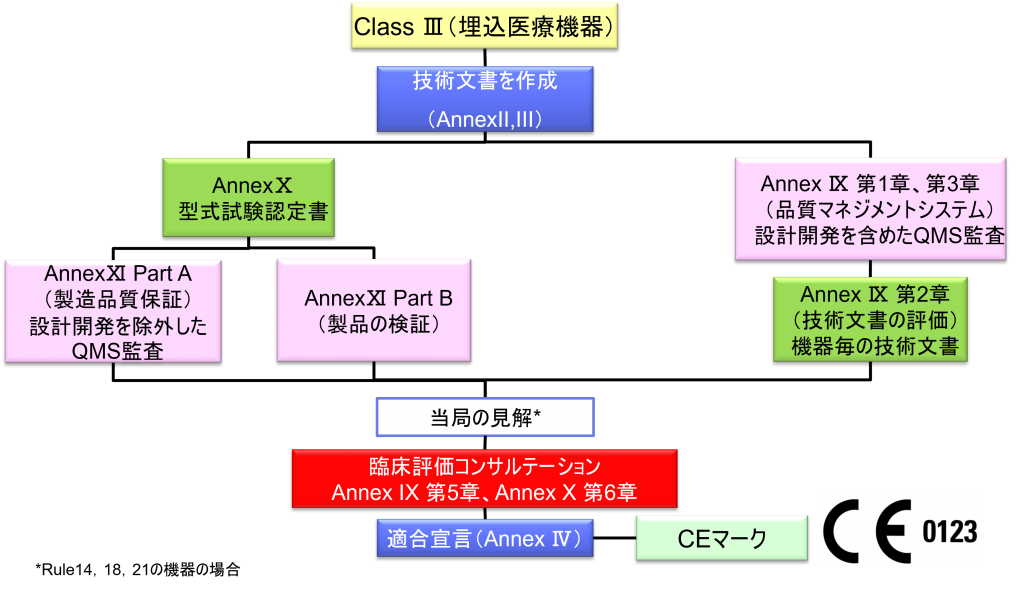
Class Ⅲ埋込医療機器の適合性評価ルート
お役立ち動画
世界一わかりやすいMDRセミナー
【第1講】欧州制度概要
【第2講】MDR概要
【第3講】規則実行に関連する機関等
【第4講】移行スケジュール
【第5講】MDRの要点
【第6講】用語の定義
【第7講】概要製造業者の責務
【第8講】規制遵守責任者
【第9講】一般的なMDR対応の流れ
【第10講】MDRが適用される機器・製品
【第11講】クラス分類
【第12講】安全性および性能の要求事項
【第13講】技術文書
【第14講】臨床評価
【第15講】 適合性評価手順
【第16講】欧州指定代理人
【第17講】輸入業者
【第18講】販売業者
【第19講】UDI
【第20講】EUDAMEDへの登録
関連商品
[blogcard url=”https://ecompliance.co.jp/SHOP/O063.html” title=”MDR(欧州医療機器規則)施行直前セミナー” content=”いよいよ5月26日にMDR(欧州医療機器規則)が完全施行されます。
品目認証の有効期間に関わらず、5月26日以降はPMSおよびビジランスはMDRの要求を満たす必要があります。
また、クラスⅠ機器でNBの関与を必要としない機器に関しては、2021年5月26日からMDRを遵守する必要があります。
しかしながら、MDRは行政側の準備が整っていない面もあります。
たとえば、経済事業者の情報、UDI情報、安全性情報等を登録するためのデータベースであるEUDAMEDはまだ完全稼働していません。
この点に関しては、MDCGがMDCG 2021-1「EUDAMEDが完全に機能するまでの、調和のとれた管理慣行と代替技術ソリューションに関するガイダンス」(Guidance on harmonised administrative practices and alternative technical solutions until EUDAMED is fully functional)を発出しています。
EUDAMEDが稼働するまでの代替プロセスが解説されています。
本セミナーでは、MDR完全施行に伴い、医療機器企業が準備しなければならない事項を分かりやすく解説します。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/EL-080.html” title=”【セミナービデオ】MDR(欧州医療機器規則)施行直前セミナー” content=”いよいよ5月26日にMDR(欧州医療機器規則)が完全施行されます。
品目認証の有効期間に関わらず、5月26日以降はPMSおよびビジランスはMDRの要求を満たす必要があります。
また、クラスⅠ機器でNBの関与を必要としない機器に関しては、2021年5月26日からMDRを遵守する必要があります。
しかしながら、MDRは行政側の準備が整っていない面もあります。
たとえば、経済事業者の情報、UDI情報、安全性情報等を登録するためのデータベースであるEUDAMEDはまだ完全稼働していません。
この点に関しては、MDCGがMDCG 2021-1「EUDAMEDが完全に機能するまでの、調和のとれた管理慣行と代替技術ソリューションに関するガイダンス」(Guidance on harmonised administrative practices and alternative technical solutions until EUDAMED is fully functional)を発出しています。
EUDAMEDが稼働するまでの代替プロセスが解説されています。
本セミナーでは、MDR完全施行に伴い、医療機器企業が準備しなければならない事項を分かりやすく解説します。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/L_MDR.html” title=”欧州医療機器規則MDR(Medical Device Regulation)セミナー” content=”2017年5月5日に、医療機器指令 Medical Device Directive (93/42/EEC)が改正され 「欧州医療機器規則 (MDR:Medical Device Regulation)」 が公示されました。
20日後の2017年5月25日より施行され、2020年5月25日までの3年間が移行措置期間とされています。Manufacturer は、この移行期間中に技術文書を改訂し、新しい要求事項に対応しなければなりません。
MDRは、従来のMDDと能動埋め込み型医療機器指令 Active Implantable Medical Device Directive (90/385/EEC)を統合し置き換えるものです。
MDDからMDRへの改正内容は多岐に渡り、また上市済の製品の対策も必要となるため、強制化に間に合うように計画的に対策を進める必要があります。
MDRは、日本や米国の法規制を参考にして作成され、世界で最も厳しい規制となりました。
しかしながらMDRは条文が難解で非常にわかりにくいのが難点です。
いったい何が要求されどのように対応したら良いのでしょうか?
MDRは、技術文書について、従来よりも踏み込んだ精査を求めています。
また臨床評価と市販後臨床フォローアップについてより厳しい要求事項となりました。
本セミナーでは、MDRの要点をわかりやすく解説し、MDDからの変更点を明らかにします。また優先すべき事項、対応のための問題点・リスクを解説します。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/EL-028.html” title=”【セミナービデオ】欧州医療機器規則MDR(Medical Device Regulation)セミナー
” content=”2017年5月5日に、医療機器指令 Medical Device Directive (93/42/EEC)が改正され 「欧州医療機器規則 (MDR:Medical Device Regulation)」 が公示されました。
20日後の2017年5月25日より施行され、2020年5月25日までの3年間が移行措置期間とされています。Manufacturer は、この移行期間中に技術文書を改訂し、新しい要求事項に対応しなければなりません。
MDRは、従来のMDDと能動埋め込み型医療機器指令 Active Implantable Medical Device Directive (90/385/EEC)を統合し置き換えるものです。
MDDからMDRへの改正内容は多岐に渡り、また上市済の製品の対策も必要となるため、強制化に間に合うように計画的に対策を進める必要があります。
MDRは、日本や米国の法規制を参考にして作成され、世界で最も厳しい規制となりました。
しかしながらMDRは条文が難解で非常にわかりにくいのが難点です。
いったい何が要求されどのように対応したら良いのでしょうか?
MDRは、技術文書について、従来よりも踏み込んだ精査を求めています。
また臨床評価と市販後臨床フォローアップについてより厳しい要求事項となりました。
本セミナーでは、MDRの要点をわかりやすく解説し、MDDからの変更点を明らかにします。また優先すべき事項、対応のための問題点・リスクを解説します。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/200716-P.html” title=”欧州医療機器規則(MDR)におけるPMS・ビジランス対応セミナー” content=”コロナ禍によりMDR(Medical Device Regulation:欧州医療機器規則)の完全施行が2021年5月まで延期となりました。
日本の企業はMDRの全貌を適切に理解していないと思われ、対応が後手に回っていると思われます。
MDRは、FDAや日本の規制要件、および医薬品の規制要件などを参考に構築されており、いわば世界一厳しい医療機器規制要件となりました。
とりわけ、市販後監視(PMS)およびビジランスシステムに関しては難解であり、適切に理解している企業は稀であると言っても過言ではないでしょう。
本邦においては、QMS省令とGVP省令は明確に区別されていますが、MDRにおいては、PMS、ビジランスの要求事項もQMSに取り入れて構築する必要があります。
また当該QMSは欧州の手順のみを記載すれば良いのではなく、日本や米国など欧州圏外のPMSやビジランスの手順をすべて含めることとされています。
また市販後監視に関わる技術文書の要求事項が新設され、市販後監視計画書なども技術文書として管理・維持することが求められています。
本セミナーでは、医薬品GVPを熟知した講師が、欧州医療機器規則におけるPMSおよびビジランスに関して分かりやすく解説を行います。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/EL-039.html” title=”【セミナービデオ】欧州医療機器規則(MDR)におけるPMS・ビジランス対応セミナー” content=”コロナ禍によりMDR(Medical Device Regulation:欧州医療機器規則)の完全施行が2021年5月まで延期となりました。
日本の企業はMDRの全貌を適切に理解していないと思われ、対応が後手に回っていると思われます。
MDRは、FDAや日本の規制要件、および医薬品の規制要件などを参考に構築されており、いわば世界一厳しい医療機器規制要件となりました。
とりわけ、市販後監視(PMS)およびビジランスシステムに関しては難解であり、適切に理解している企業は稀であると言っても過言ではないでしょう。
本邦においては、QMS省令とGVP省令は明確に区別されていますが、MDRにおいては、PMS、ビジランスの要求事項もQMSに取り入れて構築する必要があります。
また当該QMSは欧州の手順のみを記載すれば良いのではなく、日本や米国など欧州圏外のPMSやビジランスの手順をすべて含めることとされています。
また市販後監視に関わる技術文書の要求事項が新設され、市販後監視計画書なども技術文書として管理・維持することが求められています。
本セミナーでは、医薬品GVPを熟知した講師が、欧州医療機器規則におけるPMSおよびビジランスに関して分かりやすく解説を行います。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/200729-P.html” title=”欧州医療機器規則(MDR)における臨床評価セミナー” content=”コロナ禍によりMDR(Medical Device Regulation:欧州医療機器規則)の完全施行が2021年5月まで延期となりました。
日本の企業はMDRの全貌を適切に理解していないと思われ、対応が後手に回っていると思われます。
MDRは、FDAや日本の規制要件、および医薬品の規制要件などを参考に構築されており、いわば世界一厳しい医療機器規制要件となりました。
とりわけ、MDRでは臨床評価要求事項(臨床的な安全評価・有効性評価)が厳格化されました。
臨床評価はMDD(Annex X)でも要求されていましたが、MDRにおいても臨床評価を実施し、臨床評価報告書を作成する必要があります。
臨床評価は機器クラスや新規性とは関係なく、全ての機器に必要であり、日本の要求と異なることの1つです。
実施のための詳細は臨床評価ガイダンス(MEDDEV 2.7/1 revision 4)に記載されています。
MDRの臨床評価では、ガイダンスの内容の大項目の一部を含めた上で、さらに要件の追加および変更を行っています。
臨床データと臨床評価報告書(CER)は精査し、また、これらを繰り返し更新することも要求されます。
製造業者は、MDR Article 61およびAnnex XIVにPart A(臨床評価関連のAnnex)に基づいて臨床評価と市販後臨床フォローアップ(PMCF)を実施しなければなりません。
製造業者は臨床評価レベルの適切性について正当化することが求められます。
臨床評価は下記に基づく必要があります。
a)科学的文献(Scientific Literature)
b)臨床試験(Clinical Investigation)
c)他のオプションとなる治療法の考慮(Other treatment)
当然のことながら、認証機関の審査する技術資料の1つとして臨床評価が含まれます。
臨床評価コンサルテーションも導入され、リスクの高い医療機器について、臨床評価の審査に行政当局が介入することになりました。
それに伴い、臨床試験実施可否判断基準も厳格化されました。
MDRにおいてはリスクの高い医療機器は、臨床試験が原則的に必須です。
リスクの高い医療機器は、他社の医療機器との同等性を示す場合、その医療機器の技術文書へのフルアクセスの契約があることが条件となりますが、事実上は難しいでしょう。
埋め込み機器およびクラス?機器の場合は、安全性と臨床性能のサマリー(SSCP:Summary of Safety and Clinical Performance)の作成が求められます。
臨床評価は市販後調査(Post-Market Surveillance)および市販後臨床フォローアップ(Post-Market Clinical Follow-up)とも関連します。
市販後監視で集めた情報によって臨床評価を更新することも忘れてはなりません。
PMCFのより詳細な考察や、定期的な安全性アップデート報告書(クラス?a以上の機器)の提出が求められます。
本セミナーでは、日米欧の医療機器規制要件を熟知した講師が、欧州医療機器規則における臨床評価に関して分かりやすく解説を行います。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/EL-040.html” title=”【セミナービデオ】欧州医療機器規則(MDR)における臨床評価セミナー” content=”コロナ禍によりMDR(Medical Device Regulation:欧州医療機器規則)の完全施行が2021年5月まで延期となりました。
日本の企業はMDRの全貌を適切に理解していないと思われ、対応が後手に回っていると思われます。
MDRは、FDAや日本の規制要件、および医薬品の規制要件などを参考に構築されており、いわば世界一厳しい医療機器規制要件となりました。
とりわけ、MDRでは臨床評価要求事項(臨床的な安全評価・有効性評価)が厳格化されました。
臨床評価はMDD(Annex X)でも要求されていましたが、MDRにおいても臨床評価を実施し、臨床評価報告書を作成する必要があります。
臨床評価は機器クラスや新規性とは関係なく、全ての機器に必要であり、日本の要求と異なることの1つです。
実施のための詳細は臨床評価ガイダンス(MEDDEV 2.7/1 revision 4)に記載されています。
MDRの臨床評価では、ガイダンスの内容の大項目の一部を含めた上で、さらに要件の追加および変更を行っています。
臨床データと臨床評価報告書(CER)は精査し、また、これらを繰り返し更新することも要求されます。
製造業者は、MDR Article 61およびAnnex XIVにPart A(臨床評価関連のAnnex)に基づいて臨床評価と市販後臨床フォローアップ(PMCF)を実施しなければなりません。
製造業者は臨床評価レベルの適切性について正当化することが求められます。
臨床評価は下記に基づく必要があります。
a)科学的文献(Scientific Literature)
b)臨床試験(Clinical Investigation)
c)他のオプションとなる治療法の考慮(Other treatment)
当然のことながら、認証機関の審査する技術資料の1つとして臨床評価が含まれます。
臨床評価コンサルテーションも導入され、リスクの高い医療機器について、臨床評価の審査に行政当局が介入することになりました。
それに伴い、臨床試験実施可否判断基準も厳格化されました。
MDRにおいてはリスクの高い医療機器は、臨床試験が原則的に必須です。
リスクの高い医療機器は、他社の医療機器との同等性を示す場合、その医療機器の技術文書へのフルアクセスの契約があることが条件となりますが、事実上は難しいでしょう。
埋め込み機器およびクラス?機器の場合は、安全性と臨床性能のサマリー(SSCP:Summary of Safety and Clinical Performance)の作成が求められます。
臨床評価は市販後調査(Post-Market Surveillance)および市販後臨床フォローアップ(Post-Market Clinical Follow-up)とも関連します。
市販後監視で集めた情報によって臨床評価を更新することも忘れてはなりません。
PMCFのより詳細な考察や、定期的な安全性アップデート報告書(クラス?a以上の機器)の提出が求められます。
本セミナーでは、日米欧の医療機器規制要件を熟知した講師が、欧州医療機器規則における臨床評価に関して分かりやすく解説を行います。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/MD-QMS-169.html” title=”【MDR対応QMSひな形】サプライチェーン管理手順書” content=”CEマーキングを貼付し、欧州に医療機器を輸出している医療機器企業は、MDDに置き換わって2021年5月25日までに欧州医療機器規則(MDR:Medical Device Regulation)に対応する必要があります。
製造業者(Manufacturer)は、この移行期間中に技術文書を改訂し、新しい要求事項に対応しなければなりません。
MDRは、従来のMDDと能動埋め込み型医療機器指令 Active Implantable Medical Device Directive (90/385/EEC)を統合し置き換えるものです。
MDDからMDRへの改正内容は多岐に渡り、また上市済の製品の対策も必要となるため、強制化に間に合うように計画的に対策を進める必要があります。
MDRは、日本や米国の法規制を参考にして作成され、世界で最も厳しい規制となりました。
しかしながらMDRは条文が難解で非常にわかりにくいのが難点です。
本邦におけるQMS省令や米国FDAのQSRにはない要求事項も含まれます。
そのため、多くのQMSに対して加筆修正が発生し、また追加で作成しなければならないQMSもあります。
本MDR対応QMSひな形を使用することで、より短時間で品質良くMDRに対応したQMSを構築することが出来ます。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/MD-QMS-168.html” title=”【MDR対応QMSひな形】欧州指定代理人用手順書” content=”CEマーキングを貼付し、欧州に医療機器を輸出している医療機器企業は、MDDに置き換わって2021年5月25日までに欧州医療機器規則(MDR:Medical Device Regulation)に対応する必要があります。
製造業者(Manufacturer)は、この移行期間中に技術文書を改訂し、新しい要求事項に対応しなければなりません。
MDRは、従来のMDDと能動埋め込み型医療機器指令 Active Implantable Medical Device Directive (90/385/EEC)を統合し置き換えるものです。
MDDからMDRへの改正内容は多岐に渡り、また上市済の製品の対策も必要となるため、強制化に間に合うように計画的に対策を進める必要があります。
MDRは、日本や米国の法規制を参考にして作成され、世界で最も厳しい規制となりました。
しかしながらMDRは条文が難解で非常にわかりにくいのが難点です。
本邦におけるQMS省令や米国FDAのQSRにはない要求事項も含まれます。
そのため、多くのQMSに対して加筆修正が発生し、また追加で作成しなければならないQMSもあります。
本MDR対応QMSひな形を使用することで、より短時間で品質良くMDRに対応したQMSを構築することが出来ます。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/MD-QMS-152.html” title=”【FDA CFR 803対応】MDR手順書” content=”最近、MDR(有害事象報告)に関する指摘が多く出されるようになってきました。 FDAへの
有害事象(事故)報告は、所定の期間内に提出しなければなりません。”]
[blogcard url=https://xn--2lwu4a.jp/qms-md/ title=”QMS(手順書)ひな形 医療機器関連” ]
Thanks for any other informative site. Where else
may I am getting that kind of information written in such an ideal means?
I have a venture that I am simply now running on, and I’ve been at the glance out for such information.
unekwugub cssok kivmgfk lejn fvqviwqoeqtkqms
602 896778Some genuinely good and beneficial information on this site , besides I believe the style contains wonderful capabilities. 640651
895034 585618There couple of fascinating points at some point in this posting but I dont determine if these individuals center to heart. There is some validity but Let me take hold opinion until I check into it further. Fantastic write-up , thanks and then we want a lot more! Combined with FeedBurner in addition 6542
843377 496211Not long noticed concerning your web internet site and are nonetheless already reading along. I assumed ill leave my initial comment. i do not verify what saying except that Ive enjoyed reading. Good weblog. ill be bookmarking maintain visiting this internet web site truly typically. 64089
143854 782202I like this site its a master peace ! Glad I detected this on google . 441448
910573 909759The when I just read a blog, Im hoping that this doesnt disappoint me approximately this 1. Get real, Yes, it was my method to read, but When i thought youd have something interesting to state. All I hear is really a number of whining about something which you could fix really should you werent too busy trying to locate attention. 264402
919721 624899How can I attract far more hits to my composing weblog? 103934
978606 618003Some truly nice stuff on this site , I like it. 667122
288047 300788Some actually wondrous function on behalf with the owner of this site, perfectly excellent subject material . 28393
44857 66232Id always want to be update on new articles on this web internet site , saved to favorites ! . 401569
477107 576068extremely good post, i surely enjoy this fabulous website, persist with it 825463
288146 840332Wohh just what I was searching for, appreciate it for putting up. 621855
424087 786755Im having slightly issue I cant subscribe your feed, Im utilizing google reader fyi. 339650
183288 348048Effectively worded post will be sharing this with my readers this evening 65565
167427 780364Some truly amazing content material on this web internet site , appreciate it for contribution. 999090
587896 322572Exceptional read, I just passed this onto a colleague who was performing just a little research on that. And he in fact bought me lunch because I discovered it for him smile So let me rephrase that. 168626
722590 978546One can undertake all sorts of advised excursions with assorted limousine functions. Various offer wonderful courses and numerous can take clients for just about any ride your bike over the investment banking region, or even for a vacation to new york. ??????? 880677
749890 721617I found your weblog internet web site on google and check some of your early posts. Continue to sustain up the superb operate. I just further up your RSS feed to my MSN News Reader. Looking for forward to studying extra from you in a whilst! 837021
642627 668692I like your writing style genuinely loving this internet internet site . 801929
526808 349914I like this internet weblog really much so significantly superb info . 789249
233306 134392Do you wear boxers or biefs? I wana bui em. 340406
858681 558941Hey! Nice stuff, do tell us when you post something like that! 230434
372485 580319I consider something truly special in this internet site. 22123
916336 889444But wanna say that this is invaluable , Thanks for taking your time to write this. 520238
882647 66533Thanks – Enjoyed this post, can you make it so I receive an email when you make a fresh post? From Online Shopping Greek 565986
576674 702173Some genuinely nice and utilitarian info on this web site , likewise I think the layout has great attributes. 33581
364996 566732never saw a internet site like this, relaly impressed. compared to other blogs with this post this was definatly the best website. will save. 747774
744269 258682You completed certain good points there. I did looking on the topic matter and found most persons will go together together with your weblog 64462
630543 488594I very happy to uncover this internet site on bing, just what I was seeking for : D too bookmarked . 422560
281934 526598I truly like your writing style, excellent info , thankyou for putting up : D. 173144
350044 298496This is really fascinating, Youre a extremely skilled blogger. Ive joined your rss feed and appear forward to seeking much more of your magnificent post. Also, Ive shared your internet website in my social networks! 391136
377005 180566Even though youre any of the lucky enough choices, it comes evidently, although capture the fancy of the particular coveted by ly folks other useful you you meet might possibly properly have hard times this specific dilemma. pre owned awnings 63
Hi there, simply was aware of your weblog thru Google, and located that it is truly informative. I’m gonna watch out for brussels. I’ll appreciate for those who continue this in future. A lot of other people can be benefited from your writing. Cheers!
Thank you for sharing excellent informations. Your website is very cool. I am impressed by the details that you have on this blog. It reveals how nicely you perceive this subject. Bookmarked this web page, will come back for more articles. You, my friend, ROCK! I found simply the information I already searched everywhere and just could not come across. What a great web-site.
Nice post. I be taught something more challenging on completely different blogs everyday. It can at all times be stimulating to read content from different writers and follow just a little something from their store. I’d desire to use some with the content on my weblog whether or not you don’t mind. Natually I’ll give you a link in your net blog. Thanks for sharing.
I visited a lot of website but I conceive this one has got something special in it in it
naturally like your web site however you need to take a look at the spelling on several of your posts. A number of them are rife with spelling problems and I find it very bothersome to tell the truth on the other hand I will surely come again again.
Some truly fantastic info , Gladiola I found this. “If you find it in your heart to care for somebody else, you will have succeeded.” by Maya Angelou.
Pretty element of content. I just stumbled upon your site and in accession capital to assert that I acquire actually enjoyed account your weblog posts. Anyway I’ll be subscribing in your augment and even I fulfillment you access consistently rapidly.
I carry on listening to the news talk about getting free online grant applications so I have been looking around for the most excellent site to get one. Could you advise me please, where could i find some?
you’re really a good webmaster. The web site loading speed is amazing. It seems that you are doing any unique trick. Furthermore, The contents are masterpiece. you have done a magnificent job on this topic!
Woah! I’m really digging the template/theme of this blog. It’s simple, yet effective. A lot of times it’s hard to get that “perfect balance” between superb usability and visual appearance. I must say you have done a excellent job with this. Additionally, the blog loads super fast for me on Opera. Outstanding Blog!
Im now not positive where you are getting your information, but great topic. I needs to spend some time finding out much more or figuring out more. Thanks for wonderful info I was in search of this information for my mission.
Great blog here! Also your website loads up fast! What web host are you using? Can I get your affiliate link to your host? I wish my site loaded up as fast as yours lol
Its wonderful as your other content : D, appreciate it for posting.
Very interesting details you have remarked, appreciate it for putting up. “The unspoken word never does harm.” by Kossuth.
Of course, what a magnificent site and revealing posts, I definitely will bookmark your site.All the Best!
I delight in, cause I found exactly what I was taking a look for.
You have ended my four day long hunt! God Bless you man. Have a nice day.
Bye
I do accept as true with all the ideas you’ve introduced to your post. They are really convincing and can definitely work. Still, the posts are very short for novices. May you please lengthen them a little from subsequent time? Thank you for the post.
This web site is really a walk-through for all of the info you wanted about this and didn’t know who to ask. Glimpse here, and you’ll definitely discover it.
Thanks for sharing superb informations. Your website is very cool. I am impressed by the details that you have on this blog. It reveals how nicely you perceive this subject. Bookmarked this website page, will come back for more articles. You, my pal, ROCK! I found just the information I already searched everywhere and just could not come across. What a perfect website.
Good – I should definitely pronounce, impressed with your website. I had no trouble navigating through all tabs as well as related information ended up being truly simple to do to access. I recently found what I hoped for before you know it at all. Quite unusual. Is likely to appreciate it for those who add forums or something, website theme . a tones way for your customer to communicate. Nice task..
Whoa! This blog looks just like my old one! It’s on a totally different topic but it has pretty much the same page layout and design. Excellent choice of colors!
I found your blog site on google and test a few of your early posts. Continue to maintain up the superb operate. I just additional up your RSS feed to my MSN Information Reader. Seeking forward to reading more from you later on!…
This actually answered my problem, thanks!
A lot of thanks for all of your work on this web site. Kate enjoys going through investigation and it’s really easy to see why. Many of us learn all regarding the dynamic tactic you make great ideas through the blog and even welcome participation from people on that idea then our simple princess is always starting to learn a lot. Take advantage of the rest of the new year. You are always performing a terrific job.
I’m really enjoying the design and layout of your blog. It’s a very easy on the eyes which makes it much more enjoyable for me to come here and visit more often. Did you hire out a designer to create your theme? Excellent work!
Helpful information. Lucky me I found your web site accidentally, and I’m stunned why this twist of fate did not came about earlier! I bookmarked it.
Chúng mày cầm tiền lừa đảo của người khác có ngủ ngon không?
Nội dung toàn xúi bậy, cổ súy cho mấy cái trò phạm pháp.
I loved up to you’ll receive performed right here. The sketch is tasteful, your authored material stylish. nevertheless, you command get bought an edginess over that you would like be turning in the following. sick unquestionably come further previously again since exactly the same nearly very regularly inside case you shield this increase.
What i don’t realize is actually how you’re no longer really much more neatly-preferred than you may be right now. You are so intelligent. You understand therefore significantly in the case of this subject, produced me for my part believe it from a lot of numerous angles. Its like men and women aren’t interested until it is something to accomplish with Girl gaga! Your own stuffs outstanding. At all times deal with it up!
Nhìn chuyên nghiệp vậy thôi chứ bên trong toàn lừa đảo, dụ dỗ đầu tư vớ vẩn.
Hugo Casino bietet ein vollwertig responsives Design, das
sich jedem Endgerät anpasst. Hugo Casino überzeugt mit
einem großzügigen Willkommensbonus und regelmäßigen Promotions
für Bestandskunden. Österreichische Nutzer finden bei Hugo Casino
viele bekannte Slots und spannende Casinospiele. Casino Hugo bietet Kundenbetreuung per Live-Chat
und E-Mail. Die Website ist von Curacao lizenziert, was eine sichere Umgebung für Spieler gewährleistet.
Hugo Casino wurde 2023 gegründet und ist ein lebendiges und ansprechendes Online-Casino, das eine breite Palette von Spielen und Aktionen bietet.
Hugo Casino bietet eine Vielzahl an attraktiven Bonusangeboten und
Aktionen, die sowohl neue als auch bestehende
Spieler begeistern. Zudem prüfen wir den Registrierungsprozess inklusive KYC-Anforderungen,
die Vielfalt der angebotenen Spiele, Zahlungswege und den Kundensupport.
Mit analytischer Präzision präsentieren wir eine Plattform,
die dank eines attraktiven Willkommensbonus und moderner Sicherheitsstandards
überzeugt.
References:
https://online-spielhallen.de/vulkanspiele-casino-2024-test-boni-im-uberblick/
Diese Spiele machen viel Spaß und sind eine der neuesten Entwicklungen in der Welt der Automatenspiele.
Es gibt auch Jackpot-Spiele, bei denen die Einsätze
aller Spieler der besten online Casinos in den Jackpot einfließen,
wodurch noch größere Summen im Jackpot landen. Außerdem gibt es bei Lucky7Even DE viele Arten von online Casino Echtgeld Spielautomaten mit unterschiedlichen Themen und Designs.
Du kannst dabei aus einer Vielzahl von Themen und Spielarten wählen, darunter Video-Slots,
progressive Jackpot-Slots, online Blackjack, online Roulette, Baccarat und mehr.
Ob Tischspiele, Live-Games, die beliebtesten Slots und vieles mehr.
Wer mobil unterwegs ist, kann in der Lucky7even Spielothek online alle Zahlungsmethoden bequem nutzen. Außerdem gibt
es bei Lucky7even viele Arten von online Casino Echtgeld Spielautomaten mit unterschiedlichen Themen und Designs.
Casinolucky7even.de ist eine Online-Ressource, die dem beliebten Casino Lucky7even gewidmet ist.
Die Freispiele und Bonusaktionen sind super attraktiv und fair gestaltet.
Ich spiele nun schon seit einiger Zeit bei
Lucky7even Casino und bin absolut zufrieden!
Mit der Glückszahl 7 im Namen bietet das Lucky7even Casino
eine umfangreiche Auswahl an Spielen an und begrüßt Neukunden mit einem umfassenden Willkommensbonus von bis zu 2.000€ und
200 Freispielen. Und dein erster Einzahlungsbonus mit 100% bis zu 500€
und 50 Freispielen ist auch nicht mehr weit entfernt. Lucky7even Casino bietet
eine große Auswahl an Spielen, von klassischen Spielautomaten und Tischspielen bis
hin zu Live-Casino-Spielen. Besucher von casinolucky7even.de sollten sich über die Gesetze und Steuern in ihrem Wohnsitzland in Bezug auf Casinospiele informieren. Lucky7Even De bietet eine große Auswahl an Spielen, von klassischen Spielautomaten und Tischspielen bis hin zu Live-Casino-Spielen. Wenn Sie nicht genug Geld haben,
um zu spielen, sollten Sie unsere Finanzierungsoptionen in Betracht
ziehen.
References:
https://online-spielhallen.de/umfassende-bewertung-der-buran-casino-mobile-app/
Web này là ổ rửa tiền, dây vào chỉ có đi tù. Né gấp!
With a wide range of pokies, table games, and more, the 21bit Casino Australia lobby is designed
for easy browsing. New players at 21bit Casino Australia can claim a AU$1,000 + 250
Free Spins Welcome Package. The platform offers more than 3,000 games,
loyalty rewards that give back, and a fair play environment.
New players can get started with a €750 + 250 Free Spins Welcome Bonus.
It is a safe and crypto-friendly destination where you will find the best slots EU, exciting live dealer tables and promotions that reward you every step of the way.
Whether you deposit in traditional currency or prefer crypto, 21bit Casino EU makes payments smooth, transparent and secure.
If you’re like most Aussie gamblers, pokies are probably your go-to.
Consider this your unofficial guide to whether 21bit is worth your hard-earned dough.
Alright, let’s get into the nitty-gritty of 21bit Casino for us Aussies.
Prepare yourself for a seriously huge library of
over 3000 pokies, covering every theme and feature you can imagine.
It boasts a massive pokies selection, offering thousands of titles to keep you spinning.
References:
https://blackcoin.co/level-up-casino-login-guide/
Playing slots with cryptocurrency is legal, even in countries where gambling is prohibited by local authorities.
But, some prefer to play bitcoin slots in which there are many different features.
New players who do not know much about slots should start
with Low Volatility slots and look at what percentage of RTP is in the slot.
By the way, volatility and RTP are the most important parameters
that a player who play slots with crypto should pay attention to.
And, in every online crypto casinos, this figure
will be identical for the same slots. Basically, all BTC slots consist of three
or more reels that rotate from top to bottom and have additional features such as bonus mini-games.
Don’t hesitate to contact them via email or live chat.
We invite you to take the best of Litecoin gambling and
Ethereum gambling to make epic wins. Our goal is to
make you feel safe while you spend your best moments with
7bit. Bitcoin is a new word in online payments. Our philosophy is simple – to make you forget about
all troubles and worries and spend a fantastic
time at our casino.
References:
https://blackcoin.co/18_what-is-a-high-roller-at-a-casino-what-high-roller-actually-means_rewrite_1/
Earn entry tickets on Table Games or on-site
NT Keno for your chance to win great prizes! The most popular
include Roaring 21, Rich Casino, Black Diamond, Slotland, and 7Bit Casino.
The best thing you can do is find a website that offers fair chances to the player, with low volatility and a high
RTP score. There really isn’t a simple trick that you can use to win.
Instead, you can play them via your desktop or mobile browser.
When it comes to slots, there are loads, including favorites like Starburst, Gonzo’s Quest and Game of Thrones.
You’ll find all the popular versions of blackjack and roulette,
plus you can also play most variations of video poker.
Certain games, such as blackjack, may require an element of
strategy in order to win. Enter the mobile casino – the no waiting platform par excellence.
As well as becoming ever more popular, mobile gaming allows quick, short
gaming sessions. That’s why mobile gaming continues to grow in popularity.
As your phone or tablet are almost always with you, the mobile casino is pure
entertainment and fun – on the go! The rate at which Comp Points
accumulate is so fast that you can extend a gaming session for a long time by converting your Comp
Points into casino credits.
References:
https://blackcoin.co/37_best-online-casinos-for-vip-players-2022_rewrite_1/
Our casino experts have outlined three important steps
to help ensure you have a great time playing online.
Choose how you want to deposit money into your casino account and
make your first deposit. Now, you can play these games on mobile
devices like laptops, tablets, or smartphones. If you don’t find a review for an Australian online casino, feel
free to send us an email to [email protected], and
we’ll consider reviewing it in the future.
Although players could access a real money online casino in Australia, there
was no law regulating the provision of such services.
This guide explains the differences between these sites and regular casinos, how
to play pokies and other casino games online, as well as how to
cash out your winnings. A top Australian casino online in 2025 offers a
wide range of games at the click of a mouse, and it takes
just minutes to sign up for a real money casino Australia account.
By reviewing hundreds of sites of we’ve managed to rank the best online casinos for Australian players.
It wasn’t until now that I had to test it extensively and compare it with other
Australian online casinos for real money
to give it a proper ranking.
Jackpots, instant games, table games, and live
dealers are also on the repertoire, though the choices are more limited.
If you’re a high roller, though, you can deposit A$500 or more and unlock the 100% up to A$3,000 bonus.
The minimum deposit required is pretty low at
A$25, but it’s important to note the 40x wagering requirements attached.
While we weren’t too happy that the RTP isn’t clearly
displayed as information, our testers confirmed that every
game had been third-party certified.
paypal casinos online that accept
References:
spechrom.com
online slots uk paypal
References:
https://www.mvacancy.com/companies/online-casinos-that-accept-paypal-2025-nj-com/
paypal online casino
References:
http://www.s-golflex.kr/main/bbs/board.php?bo_table=free&wr_id=4892337
Keep functioning ,remarkable job!
online real casino paypal
References:
https://sosjob.ca/employer/the-10-most-popular-online-casino-games/
When I initially commented I clicked the -Notify me when new comments are added- checkbox and now each time a remark is added I get four emails with the identical comment. Is there any manner you possibly can take away me from that service? Thanks!
Some genuinely choice blog posts on this website , bookmarked.
Excellent read, I just passed this onto a colleague who was doing some research on that. And he just bought me lunch because I found it for him smile Thus let me rephrase that: Thanks for lunch!
Hey very nice blog!! Man .. Excellent .. Amazing .. I will bookmark your website and take the feeds also…I’m happy to find so many useful info here in the post, we need develop more techniques in this regard, thanks for sharing. . . . . .
When I originally commented I clicked the -Notify me when new comments are added- checkbox and now each time a comment is added I get four emails with the same comment. Is there any way you can remove me from that service? Thanks!
Contact Me – Very thoughtful article. I appreciate your perspective.
My partner and I stumbled over here from a different website and thought
I might as well check things out. I like what I see so now i am following you.
Look forward to looking at your web page repeatedly.