2014年11月25日より、これまでの薬事法を一部改正した「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律」(薬機法)が施行された。
これに伴い、医療機器の製造業が許可制から登録制へと緩和される一方、製造販売業はQMS省令の対象となるなど、大きな変更が実施された。
薬機法のポイントはこちら。
本記事に関する最新情報はこちら。
製造販売業
医療機器を市場に出す事業者(製造販売業者。輸入業者も含まれる。)は、医療機器の製造販売業許可を取得することになる。
医療機器を日本国内市場に出すにあたっては、医療機器の品質が保証され、ユーザや患者、医療関係者等の安全が確保されるものでなければならない。
そのため、薬機法では、製造販売業許可の要件として、品質保証と安全管理の体制を整えることが求められている。
医療機器製造販売業許可は、「事業者」が取得する。
一法人にひとつの許可。(第1種医療機器製造販売業許可と第3種医療機器製造販売業許可を同時に持つことはない。)
複数の営業所がある場合は、総括製造販売責任者の常駐する事業所(本社等)がある都道府県知事に、許可を申請する。
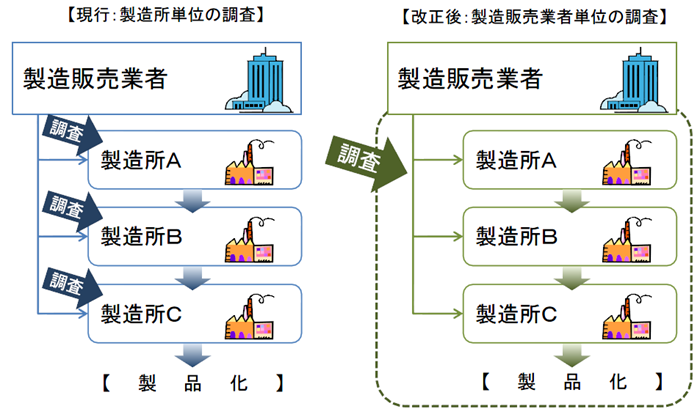
薬機法施行後は、製造販売業がQMS省令の対象となる。
従来のように製造所毎に別個に調査・判定をするのではなく、製造販売業者に対して、品質システム全体を包括的に調査・判定することになった。
製造販売業者の責任業務
製造販売業者は、次に掲げる業務を行わなければならない。
- QMSに必要な工程の内容(当該工程により達成される結果を含む。)を明らかにするとともに、当該工程のそれぞれについて、各施設の関与の態様を明確にすること。
- 工程の順序および相互の関係を明確にすること。
- 工程の実施および管理の実効性の確保に必要な判定基準および方法を明確にすること。
- 工程の実施、監視および測定に必要な資源および情報が利用できるようにすること。
- 工程を監視し、測定し、および分析すること。
- 工程について、1.の結果を得るために、および実効性を維持するために所要の措置を採ること。
体制の整備(体制省令およびGVP省令への適合)
国は、医療機器又は体外診断用医薬品の製造管理又は品質管理に係る業務を行う体制の基準(体制)、製造管理及び品質管理の基準(QMS)、医療機器の市販後安全管理に関する基準(GVP)を、省令として公布している。 医療機器の製造販売業許可業者は、これらの省令にしたがって、適切に、医療機器の品質管理・安全管理を行わねばならない。 その体制が整っていることが、「許可の取得」「許可の維持」の要件である。

製造販売業許可を取得する場合、管理監督者(経営者)、管理責任者の他に、いわゆる三役(医療機器等総括製造販売責任者、国内品質業務運営責任者、医療機器等安全管理責任者)を置かなければならない。
また製造業の登録申請を行う場合、責任技術者を置かなければならない。
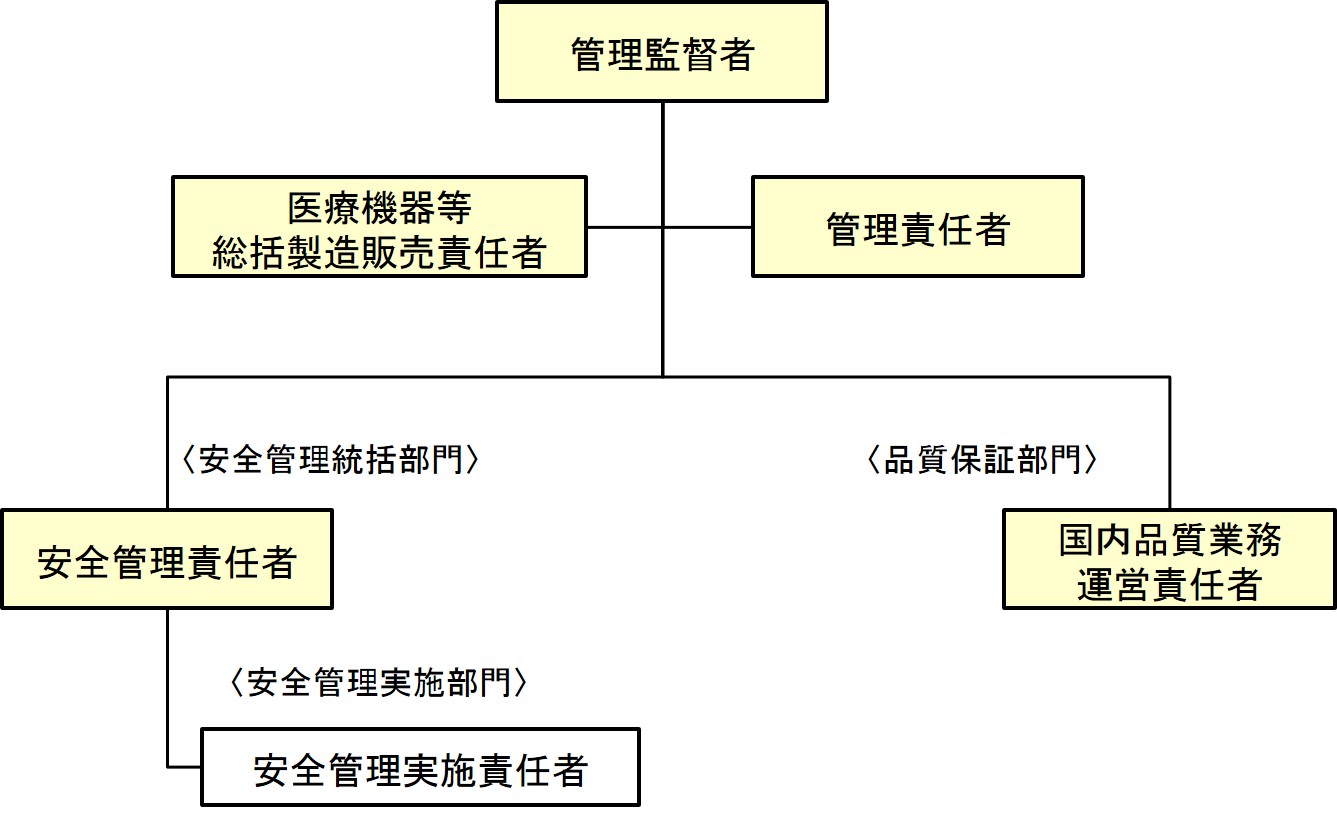
管理責任者
管理責任者は、製造販売業者等の役員、管理職の地位にある者その他これに相当する者がその任につくこと。
管理責任者の責任及び権限
- 工程が確立され、実施されるとともに、その実効性が維持されているようにすること。
- 品質管理監督システムの実施状況及びその改善の必要性について管理監督者に報告すること。
- 全ての施設において、法令の規定等及び製品受領者要求事項についての認識が向上するようにすること。
医療機器等総括製造販売責任者の業務
医療機器等総括製造販売責任者は、管理監督者、管理責任者、国内品質業務運営責任者を兼ねることができる。
- 製品の出荷の決定その他の製造管理及び品質管理に係る業務を統括し、これに責任を負うこと。
- 業務を公正かつ適正に行うために必要があると認めるときは、製造販売業者、管理監督者その他の当該業務に関して責任を有する者に対し文書により必要な意見を述べ、その写しを五年間保管すること。
- 国内品質業務運営責任者を監督すること。
- 管理責任者及び国内品質業務運営責任者(限定第三種医療機器製造販売業者にあっては、管理責任者を除く。)の意見を尊重すること。
- 安全管理統括部門との密接な連携を図らせること。
医療機器等総括製造販売責任者の要件
- クラス2以上(施行規則第114条の49第1項)高度管理医療機器又は管理医療機器の製造管理及び品質管理並びに製造販売後安全管理を行う者に係る法第23条の2の14第1項の厚生労働省令で定める基準は、次の各号のいずれかに該当する者であること。
- 大学等で物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学又は歯学に関する専門の課程を修了した者
- 旧制中学若しくは高校又はこれと同等以上の学校で、物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学又は歯学に関する専門の課程を修了した後、医薬品、医療機器又は再生医療等製品の品質管理又は製造販売後安全管理に関する業務に3年以上従事した者
- 医薬品、医療機器又は再生医療等製品の品質管理又は製造販売後安全管理に関する業務に5年以上従事した後、別に厚生労働省令で定めるところにより厚生労働大臣の登録を受けた者が行う講習を修了した者
四 厚生労働大臣が前3号に掲げる者と同等以上の知識経験を有すると認めた者
- クラス1(規則第114条の49第2項)
一般医療機器の製造管理及び品質管理並びに製造販売後安全管理を行う者に係る法第23条の2の14第1項の厚生労働省令で定める基準は、次の各号のいずれかに該当する者であることとする。
- 旧制中学若しくは高校又はこれと同等以上の学校で、物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学又は歯学に関する専門の課程を修了した者
- 旧制中学若しくは高校又はこれと同等以上の学校で、物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学又は歯学に関する科目を修得した後、医薬品、医薬部外品、化粧品、医療機器又は再生医療等製品の品質管理又は製造販売後安全管理に関する業務に3年以上従事した者
- 厚生労働大臣が前2号に掲げる者と同等以上の知識経験を有すると認めた者
国内品質業務運営責任者の業務
国内品質業務運営責任者は、管理責任者を兼ねることができる。
- 国内の品質管理業務を統括すること。
- 国内の品質管理業務が適正かつ円滑に行われていることを確認すること。
- 国内に流通させる製品について、市場への出荷の決定をロット(製造番号又は製造記号)ごとに行い、その結果及び出荷先等市場への出荷の記録を作成すること。
- 国内に流通する製品について、当該製品の品質に影響を与えるおそれのある製造方法、試験検査方法等の変更がなされる場合にあっては、当該変更に係る情報を国内外から収集し、かつ、把握するとともに、当該変更が製品の品質に重大な影響を与えるおそれがある場合には、速やかに管理責任者及び医療機器等総括製造販売責任者に対して文書により報告し、必要かつ適切な措置が採られるようにすること。
- 国内に流通する製品について、当該製品の品質等に関する情報(品質不良又はそのおそれに係る情報を含む。)を国内外から収集するとともに、当該情報を得たときは、速やかに管理責任者及び医療機器等総括製造販売責任者に対して文書により報告し、記録し、及び必要かつ適切な措置が採られるようにすること。
- 国内に流通する製品の回収を行う場合に、次に掲げる業務を行うこと。
イ 回収した医療機器等を区分して一定期間保管した後、適正に処理すること。
ロ 回収の内容を記載した記録を作成し、管理責任者及び医療機器等総括製造販売責任者に対して文書により報告すること。 - 第4号から前号までに掲げるもののほか、国内の品質管理業務の遂行のために必要があると認めるときは、管理責任者及び医療機器等総括製造販売責任者に対して文書により報告すること。
- 国内の品質管理管理業務の実施に当たり、必要に応じ、関係する登録製造所に係る製造業者又は医療機器等外国製造業者、販売業者、薬局開設者、病院及び診療所の開設者その他関係者に対し、文書による連絡又は指示を行うこと。
- 製造販売後安全管理基準第二条第二項に規定する安全確保措置に関する情報を知ったときは、安全管理統括部門に遅滞なく文書で提供すること。
国内品質業務運営責任者の要件
製造販売業者は、この省令の規定に従って行う国内の製品の品質を管理する業務の責任者として、国内に所在する施設に、次に掲げる要件を満たす国内品質業務運営責任者を置かなければならない。
- 製造販売業者における品質保証部門の責任者であること。
- 品質管理業務その他これに類する業務に三年以上従事した者であること。
- 国内の品質管理業務を適正かつ円滑に遂行しうる能力を有する者であること。
- 医療機器等の販売に係る部門に属する者でないことその他国内の品質管理業務の適正かつ円滑な遂行に支障を及ぼすおそれがない者であること。
注)国内品質業務運営責任者は、従前のQMS省令では、品質保証責任者と呼んでいた。改正法では、医薬品と医療機器を区別したため、医療機器における名称が変更になった。企業は、今後ともSOPなどで品質保証責任者と呼んでも構わない。
安全管理責任者
第一種製造販売業者は、次に掲げる要件を満たす安全確保業務の統括に係る部門(以下この章において「安全管理統括部門」という。)を置かなければならない。
- 総括製造販売責任者の監督下にあること。
- 安全確保業務を適正かつ円滑に遂行しうる能力を有する人員を十分に有すること。
- 医薬品等の販売に係る部門その他安全確保業務の適正かつ円滑な遂行に支障を及ぼすおそれのある部門から独立していること。
第一種製造販売業者は、次に掲げる要件を満たす安全確保業務の責任者(以下この章において「安全管理責任者」という。)を置かなければならない。
- 安全管理統括部門の責任者であること。
- 安全確保業務その他これに類する業務に三年以上従事した者であること。
- 安全確保業務を適正かつ円滑に遂行しうる能力を有する者であること。
- 医薬品等の販売に係る部門に属する者でないことその他安全確保業務の適正かつ円滑な遂行に支障を及ぼすおそれがない者であること。
- 第一種製造販売業者は、次項に規定する場合を除き、安全管理責任者に安全確保業務を行わせなければならない。
- 第一種製造販売業者は、安全確保業務であって規則第九十七条 各号に掲げるものの全部又は一部を安全管理責任者以外の者に行わせる場合にあっては、当該業務を適正かつ円滑に遂行しうる能力を有する当該業務の実施に係る責任者(以下「安全管理実施責任者」という。)を置かなければならない。
QMSの構築
製造販売業者は、新QMS省令にもとづき、QMSの構築を行わなければならない。

QMS適合性調査
薬機法施行後のQMS適合性調査は、製造販売業のQMSのもと、PMDAまたは登録認証機関が調査を実施する。(適合性調査を受けるのは製造販売業者+製造業者)
*QMS=企業がシステム(組織体制やルール)を確立し、製造に関わる組織全体で品質保証(製造管理及び品質管理)すること。
製造業
医療機器(ソフトウェアを含む)を製造する場合は、製造業として登録の申請(薬機法第二十三条の二の三第一項)を行う必要がある。登録の有効期限は5年間である。
医療機器やソフトウェアの設計を行う者も製造業の登録が必要であるため注意が必要である。
登録制への移行に伴い、登録申請時に添付する資料が簡素化された。
医療機器とされるソフトウェア(例:診断用ソフトウェア)を設計・開発・製造する業者も製造業の登録が必要である。経過措置として、登録対象製造所ごとに、施行日から起算して3か月以内に登録の申請をしなければならない。
すでに製造業許可を受けている製造所は、自動的に登録されるため、許可期限の残日数は何もしなくてもよい。許可更新予定日までに新たに登録することとなる。
【現行】すべて許可制 【改正後】医療機器、体外診断用医薬品は登録制へ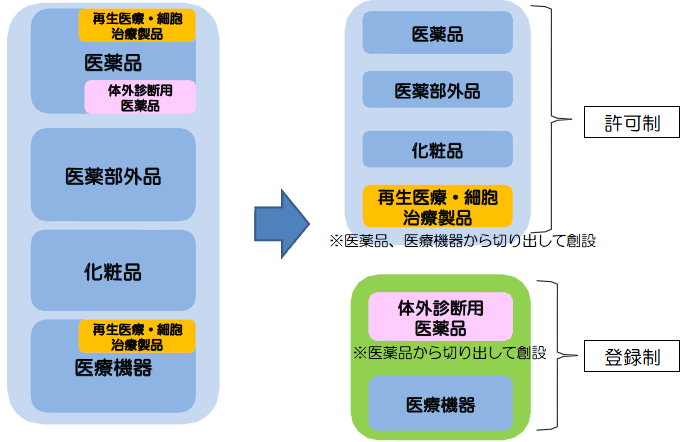
従来、許可申請時には所管都道府県によるQMS適合性調査が実施されていたが、薬機法施行後は、登録時に調査が行われることはない。
tablet for allergy on skin what is allergy medicine called names of prescription allergy pills
Asking questions are actually good thing if you are not understanding
something entirely, but this paragraph gives nice understanding yet.
My partner and I stumbled over here from a different page and thought I may as well
check things out. I like what I see so now i’m following
you. Look forward to finding out about your web page repeatedly.
Here is my web-site :: is fitspresso legit
Do you mind if I quote a couple of your articles
as long as I provide credit and sources back to your website?
My blog site is in the exact same area of interest
as yours and my visitors would really benefit from some of the information you provide here.
Please let me know if this ok with you. Thanks a lot!
my blog tonic greens facebook
This article will help the internet viewers for building up new weblog or even a weblog from start to end.
My page … the money wave scam
2pLLJS40ACVyWJkCHkkrMRxFLTf
11 Strategies To Refresh Your Pramagtic Free 프라그마틱 무료
What’s up, I log on to your blog daily. Your story-telling style is awesome, keep it up!
I read this article fully on the topic of
the resemblance of newest and previous technologies, it’s
amazing article.
Одним из современных подходов к ОЗДС является интегрированный подход, который сочетает различные методы дератизации и направлен на решение проблемы на комплексном уровне. Например, это может включать применение физических барьеров, установку ловушек, использование химических препаратов и биологических методов контроля дератов. Интегрированный подход может быть более эффективным, так как позволяет использовать сильные стороны различных методов и компенсировать их ограничения.
Good post! We will be linking to this particularly great post on our site. Keep up the great writing
Автор приводит разные аргументы и факты, позволяя читателям сделать собственные выводы.
Great information shared.. really enjoyed reading this post thank you author for sharing this post .. appreciated
There is definately a lot to find out about this subject. I like all the points you made
Мне понравилась систематическая структура статьи, которая позволяет читателю легко следовать логике изложения.
For the reason that the admin of this site is working, no uncertainty very quickly it will be renowned, due to its quality contents.
Your mode of describing everything in this piece of writing is in fact pleasant, all can easily understand it, Thanks a lot.
Good post! We will be linking to this particularly great post on our site. Keep up the great writing
30bXeYtCcocotSmOoAU9DzwwyW9
Я рад, что наткнулся на эту статью. Она содержит уникальные идеи и интересные точки зрения, которые позволяют глубже понять рассматриваемую тему. Очень познавательно и вдохновляюще!
وی ایزوله ویسلی، پودری با 6 گرم BCAA و 14 گرم
EAA در هر سروینگ است که با روش میکروفیلتراسیون جریان
متقاطع تولید میشود.
پروتئین وی ایزوله، دارای پروتئین بالا و چربی و کربوهیدرات پایینتری نسبت به سایر انواع پروتئین است.
پروتئین وی هیدرولیز، باعث میشود تا با سرعت بیشتری به هدف موردنظرکه اندامی خوش فرم است برسید.
پروتئین وی، باعث میشود تا با سرعت بیشتری به هدف موردنظرکه اندامی خوش فرم است برسید.
فیتنس مکمل، دارای بهترین مکمل های خارجی و اورجینال، شامل پروتئین وی و…
پروتئین کازئین rule 1، مکملی ایدهآل برای تأمین پروتئین در طولانیمدت است. هر وعده از این مکمل، ۲۵ گرم پروتئین میسلار کازئین باکیفیت را به بدن شما میرساند.
پروتئین کازئین چیست، کازئین نوعی پروتئین از گروه فسفو پروتئینهاست که به طور طبیعی در شیر پستانداران وجود دارد.
وی نیتروتک گلد کیسه ای ماسل تک 4 کیلویی، یک مکمل پروتئینی پیشرفته است که از ترکیب منحصربهفردی از پپتیدها و ایزوله پروتئین وی تشکیل شده است.
وی ایزوله کور فا کیسه ای 500 گرمی، حاوی مقادیر قابل توجهی از آمینواسیدهای شاخهدار (BCAAs) و گلوتامین است که نقش کلیدی در ترمیم و رشد عضلات دارند.
پروتئین کازئین پلاتینیوم ماسل تک کیسهای ۲ کیلویی، مکمل پروتئینی پیشرفته و چندمنظوره است که به طور خاص برای ورزشکاران طراحی شده است.
وی ایزوله کلیر اپلاید نوتریشن کیسه ای 875 گرمی، محصولی متفاوت و باکیفیت برای ورزشکارانی است که به دنبال راهی سبک و مؤثر برای تأمین پروتئین مورد نیاز بدن خود هستند.
وی کوامترکس کیسه ای 900 گرمی، یک مکمل پودری باکیفیت است که از کنسانتره پروتئین وی ساخته شده.
وی ایزوله ایزو XP اپلاید نوتریشن کیسه ای 1 کیلویی، یک مکمل پروتئینی وی ایزوله خالص و باکیفیت است که برای رشد و حفظ عضلات بدون چربی طراحی شده.
وی رول وان کیسه ای 4500 گرمی، یک مکمل پروتئینی باکیفیت و کامل است که برای حمایت از رشد و ریکاوری عضلات طراحی شده.
وی کریتیکال اپلاید نوتریشن 900 گرمی، مکملی جامع است که علاوه بر تأمین پروتئین، به بهبود کلی عملکرد ورزشی و سلامت شما کمک میکند.
وی ایزوله رول وان کیسهای 4500 گرمی، به دلیل کیفیت بالا و طعم فوقالعادهاش، یکی از محبوبترین مکملهای پروتئینی در جهان است.
وی ایمپکت مای پروتئین کیسه ای 2500 گرمی، مکملی پرفروش و باکیفیت است که با ترکیب ویژهای از پروتئینهای باکیفیت، اسیدهای آمینه شاخهدار (BCAA) و گلوتامین، برای حمایت از عملکرد ورزشی شما طراحی شده است.
چاپ افست کارت pvc، یکی از تکنیکهای چاپ کارت پی وی سی در تیراژ بالا است که در چاپخانه و توسط دستگاههای بسیار پیشرفته صورت میگیرد.
Way cool! Some very valid points! I appreciate you writing this article plus the rest of the site is also really good.
Это позволяет читателям самостоятельно оценить представленную информацию и сделать информированные выводы.
وی دایت اپلاید نوتریشن 1 کیلویی، یکی از محبوبترین مکملهای پروتئینی در اروپا است که به دلیل ترکیب منحصربهفرد و کیفیت بالای خود شناخته میشود.
Мне понравилась систематическая структура статьи, которая позволяет читателю легко следовать логике изложения.
Please let me know if you’re looking for a article author for your blog. You have some really great posts and I think I would be a good asset. If you ever want to take some of the load off, I’d love to write some material for your blog in exchange for a link back to mine. Please shoot me an email if interested. Kudos!
وی ایزوله ایمپکت مای پروتئین کیسه ای 2.5 کیلویی، با خلوص فوقالعاده بالا، یکی از بهترین انتخابها برای ورزشکاران است.
مکمل کراتین، مکملی محبوب در دنیای بدنسازی و ورزش، ترکیبی طبیعی است که از سه اسیدآمینه آرژنین، گلایسین و متیونین در بدن تولید میشود.
مکمل پروتئین، این ماکرومغذی قدرتمند، اساس ساختار سلولها و عضلات ماست.
مکمل کراتین مونوهیدرات، یک ترکیب طبیعیه که از سه اسید آمینه گلیسین، آرژنین و متیونین ساخته میشه و به طور عمده در عضلات اسکلتی ذخیره میشه.
کراتین مونوهیدرات مای پروتئین کیسه ای 1 کیلویی، یکی از محبوبترین و بهترین مکملهای کراتین در جهان است.
Автор старается сохранить нейтральность, чтобы читатели могли основываться на объективной информации при формировании своего мнения. Это сообщение отправлено с сайта https://ru.gototop.ee/
پروتئین کوکی اپلاید نوتریشن کیسه ای 1 کیلویی، با ترکیب بینظیر پروتئین و طعم دلپذیر خمیر کوکی، تجربهای کاملاً جدید و لذتبخش را برای شما به ارمغان میآورد.
تعمیر پرینتر کارت، کارشناسان ما سعی میکنند مشکل دستگاه پرینتر شما را در کوتاهترین زمان ممکن شناسایی و آن را حل نمایند.
کراتین مونوهیدرات اینر ارمور 400 گرمی، میتواند به شما در ریکاوری و بهبود عملکرد ورزشی کمک کند.
مکمل ویتامین، مواد حیاتی ای است که بدن ما برای عملکرد صحیح به آنها نیاز دارد.
مولتی ویتامین، مکملهایی هستند که ترکیبی از ویتامینها و مواد معدنی ضروری را در یک قرص یا کپسول گرد هم میآورند.
مولتی ویتامین یو اس ان 60 عددی، برای افرادی طراحی شده که سبک زندگی فعالی دارند و به دنبال تأمین کامل نیازهای روزانه خود به ویتامینها و مواد معدنی هستند.
وی اینر ارمور، از پروتئین گاوهای علفخوار نیوزلندی تهیه شده و سرشار از لوسین، یکی از آمینو اسیدهای شاخهای (BCAA)، است.
وی ایزوله زومد لبز، یک مکمل با کیفیت بالا است که از آبپنیر ایزوله با خلوص ۹۰–۹۵٪ تهیه شده و تقریبا فاقد چربی و لاکتوز است.
مولتی ویتامین A-Z مای ویتامینز، یک مکمل روزانه کامل است که با ۹۰ کپسول، نیازهای بدن شما به ویتامینها، مواد معدنی، آنتیاکسیدانها و ریزمغذیها را پوشش میدهد
کراتین چیست؟، کراتین یک ترکیب طبیعی است که در بدن انسان تولید میشود و نقش کلیدی در تأمین انرژی سریع و قدرتمند برای عضلات ایفا میکند.
وی موتانت کیسه ای 2300 گرمی، با فرمولاسیون پیشرفته و ترکیبات دقیق، یک مکمل کامل برای حمایت از رشد عضلات و بهبود عملکرد ورزشی است.
Статья обладает нейтральным тоном и представляет различные точки зрения. Хорошо, что автор уделил внимание как плюсам, так и минусам рассматриваемой темы.
کراتین مونوهیدرات میکرونایز اپتیموم نوتریشن 600 گرمی، یک مکمل باکیفیت و مؤثر برای ورزشکاران است که به افزایش قدرت و حجم عضلات کمک میکند.
لیبل پرینتر، با چاپگر معمولی فرق دارند و از مکانیزمهای خاص خود برای چاپ استفاده میکنند.
مولتی ویتامین اپتی من اپتیموم نوتریشن 90 عددی، یک مولتی ویتامین جامع و قدرتمند است که به طور اختصاصی برای نیازهای تغذیهای آقایان، به ویژه ورزشکاران، طراحی شده است.
پروتئین کازئین میسلار ناترند 2300 گرمی، به دلیل جذب آهسته و پایدار، به عضلات شما اجازه میدهد تا ساعتها از فواید اسیدهای آمینه بهرهمند شوند.
Хорошо, что автор статьи предоставляет информацию без сильной эмоциональной окраски.
وی ایزوله ایزوجکت ایوژن 900 گرمی، یک پروتئین وی ایزوله با خلوص فوقالعاده بالاست که توسط شرکت معتبر Evogen Nutrition تولید میشود.
کراتین مونوهیدرات ویکتور مارتینز 300 گرمی، نقشی حیاتی در تأمین انرژی مورد نیاز عضلات ایفا میکند.
مولتی ویتامین موتانت 60 عددی، یک مکمل جامع و قدرتمند است که بهطور خاص برای نیازهای ورزشکاران و بدنسازان طراحی شده است.
پروتئین هگزا پرو المکس 2300 گرمی، یک مکمل پروتئینی پیشرفته و باکیفیت است که برای تغذیه طولانیمدت عضلات ورزشکاران طراحی شده است.
ریبون پرینتر، اصلی ترین و مهم ترین مواد مصرفی دستگاه چاپ کارت PVC است.
کراتین مونوهیدرات ایوژن 300 گرمی، یک مکمل غذایی باکیفیت است که به طور خاص برای بهبود عملکرد ورزشی و حمایت از رشد عضلانی طراحی شده.
Я оцениваю аккуратность и точность фактов, представленных в статье.
Neat blog! Is your theme custom made or did you download it from somewhere? A theme like yours with a few simple tweeks would really make my blog jump out. Please let me know where you got your design. Many thanks
وی ایزوله یو اس ان 1800 گرمی، از برند USN، مکملی ایدهآل برای ورزشکارانی است که به دنبال بالاترین کیفیت پروتئین هستند.
کراتین مونوهیدرات ناترکس 300 گرمی، یک مکمل کراتین باکیفیت و ایمن است که برای ورزشکاران حرفهای طراحی شده.
نقش ویتامین ها در فیتنس و بدنسازی، تامین ویتامینها و مواد معدنی ضروری برای پشتیبانی از عملکرد فیزیکی طولانیمدت است.
پروتئین کازئین کوامترکس 2300 گرمی، یک مکمل پروتئینی شبانه است که برای تغذیه عضلات در طولانیمدت طراحی شده است.
مولتی ویتامین بانوان مای ویتامین، که با نام “Active Women Myvitamins” نیز شناخته میشود، یکی از محبوبترین مکملهای غذایی در بازار جهانی است.
مولتی ویتامین موتانت، یک مکمل جامع و قدرتمند است که بهطور خاص برای نیازهای ورزشکاران و بدنسازان طراحی شده است.
مولتی ویتامین اپتی من اپتیموم نوتریشن، یک مولتی ویتامین جامع و قدرتمند است که به طور اختصاصی برای نیازهای تغذیهای آقایان، به ویژه ورزشکاران، طراحی شده است.
کراتین مونوهیدرات زومد لبز 300 گرمی، یک مکمل باکیفیت و خالص است که برای ارتقاء عملکرد ورزشی طراحی شده.
وی نیترو تک ماسل تک 1 کیلویی، یک مکمل پروتئینی پیشرفته است که به طور خاص برای کمک به عضلهسازی و بهبود عملکرد ورزشی طراحی شده است.
مکمل کراتین ترکیبی، مثل یه تیم فوتبال حرفهای میمونه که هر بازیکنش یه کار خاص رو به نحو احسن انجام میده.
کراتین ترکیبی هیدراتور یو اس ان 360 گرمی، یک مکمل پیشرفته است که برای به حداکثر رساندن عملکرد ورزشی طراحی شده.
Heya just wanted to give you a quick heads up and let you know a few of the pictures aren’t loading correctly. I’m not sure why but I think its a linking issue. I’ve tried it in two different browsers and both show the same outcome.
Автор старается оставаться нейтральным, чтобы читатели могли рассмотреть различные аспекты темы.
وی ایزوله موتانت 2300 گرمی، با ارائهی ۲۵ گرم پروتئین خالص در هر پیمانه، تجربهای بینظیر از یک مکمل باکیفیت را برای شما به ارمغان میآورد.
کراتین مونوهیدرات بادی بیلدر 300 گرمی، یک فرم خالص و باکیفیت از کراتین است که توسط برند معتبر “بادی بیلدر” تولید شده است.
مولتی ویتامین مای ویتامینز دیلی 60 عددی، یک مکمل غذایی جامع و باکیفیت از برند معتبر انگلیسی مای ویتامینز (Myvitamins) است که برای مصرف روزانه طراحی شده.
Я хотел бы выразить свою благодарность автору этой статьи за исчерпывающую информацию, которую он предоставил. Я нашел ответы на многие свои вопросы и получил новые знания. Это действительно ценный ресурс!
پروتئین کوکی ادونیس 1 کیلویی، با ترکیب منحصربهفردی از وی پروتئین و پروتئین کازئین، در هر وعده ۲۱.۵ گرم پروتئین خالص را به بدن شما میرساند.
کراتین on 600 گرمی، یک مکمل باکیفیت و مؤثر برای ورزشکاران است که به افزایش قدرت و حجم عضلات کمک میکند.
وی اینر آرمور، از پروتئین گاوهای علفخوار نیوزلندی تهیه شده و سرشار از لوسین،
وی کیسه ای ماسل تک، برای ورزشکارانی طراحی شده که به دنبال بهترین نتیجه در کوتاهترین زمان ممکن هستند.
کراتین ایوژن اصل، یک مکمل غذایی باکیفیت است که به طور خاص برای بهبود عملکرد ورزشی و حمایت از رشد عضلانی طراحی شده.
کراتین مونوهیدرات استروویت 300 گرمی، یک مکمل باکیفیت و تکجزئی است که به عنوان یکی از موثرترین انواع کراتین در جهان شناخته میشود.
وی ماسل تک کیسه ای، برای ورزشکارانی طراحی شده که به دنبال بهترین نتیجه در کوتاهترین زمان ممکن هستند.
وی ایزوله ماسل تک 2300 گرمی، از برند MUSCLETECH، یک مکمل پروتئینی پیشرفته است که با استفاده از فناوریهای میکروفیلتراسیون و اولترافیلتراسیون چند فازی تولید میشود.
مولتی ویتامین الفا من مای ویتامینز 240 عددی، یک مکمل غذایی باکیفیت بالا است که به طور خاص برای نیازهای سلامت آقایان فعال طراحی شده است.
وی زو زومد لبز، محصولی فوقحرفهای و باکیفیت است که با ترکیبات استثنایی و طعمهای جذاب، انتخابی عالی برای ورزشکاران محسوب میشود.
Статья предлагает разнообразные данные и факты, представленные без пристрастия.
کراتین مونوهیدرات اسکال لبز 300 گرمی، یعنی کیفیت و خلوص! اسکال لبز (Skull Labs) یک برند لهستانیه که در تولید مکملهای ورزشی با کیفیت بالا شناخته شده.
کازئین میسلار کوامترکس 900 گرمی، یک پروتئین با جذب بسیار آهسته است که برای تأمین مداوم اسیدهای آمینه به عضلات، به ویژه در طول شب و ساعات طولانی بین وعدههای غذایی، طراحی شده است.
کراتین استروویت، یک مکمل باکیفیت و تکجزئی است که به عنوان یکی از موثرترین انواع کراتین در جهان شناخته میشود.
کراتین ایوژن، یک مکمل غذایی باکیفیت است که به طور خاص برای بهبود عملکرد ورزشی و حمایت از رشد عضلانی طراحی شده.
مولتی ویتامین سوپر ویت کوامترکس 120 عددی، یک مکمل مولتیویتامین و مولتیمینرال جامع است که توسط برند معتبر کوامترکس تولید میشود.
Статья охватывает различные аспекты обсуждаемой темы и представляет аргументы с обеих сторон.
Автор старается оставаться объективным, чтобы читатели могли сформировать свое собственное мнение на основе предоставленной информации.
I have been surfing on-line greater than three hours as of late, but I never found any fascinating article like yours. It is lovely worth enough for me. In my opinion, if all site owners and bloggers made excellent content material as you did, the web can be much more helpful than ever before.
Hello, its nice piece of writing on the topic of media print, we all understand media is a fantastic source of information.
وی گلد استاندارد اپتیموم نوتریشن 900 گرمی، با استفاده از فناوریهای فیلترینگ پیشرفته، عمدتاً از پروتئین وی ایزوله تهیه میشود که چربی، کربوهیدرات و لاکتوز اضافی آن حذف شده است.
کراتین مای پروتئین یک کیلویی، یکی از محبوبترین و بهترین مکملهای کراتین در جهان است که به دلیل خلوص و کیفیت بالا، سالهاست که در سایت ما رتبه اول را به خود اختصاص داده است.
وی ایزوله ایوژن، یک پروتئین وی ایزوله با خلوص فوقالعاده بالاست که توسط شرکت معتبر Evogen Nutrition تولید میشود.
کراتین ترکیبی ناترکس 1300 گرمی، یک محصول پیشرفته برای بارگیری گلیکوژن و کراتین است که برای به حداکثر رساندن عملکرد و حجم عضلات طراحی شده.
Статья представляет все основные аспекты темы, без излишней детализации.
Я оцениваю умение автора использовать разнообразные источники, чтобы подкрепить свои утверждения.
ماسل رولز، وی رولز پلاس ماسل رولز ترکیبی از پروتئین وی ایزوله و کنسانتره است.
وی اکستریم ناپالم فا، یک پروتئین وی کنسانتره باکیفیت و پیشرفته است که برای به حداکثر رساندن عملکرد و ریکاوری ورزشکاران طراحی شده است.
پروتئین کازئین چیست ؟، نوعی پروتئین از گروه فسفو پروتئینهاست که به طور طبیعی در شیر پستانداران وجود دارد.
مولتی ویتامین فارماتون 100 عددی، یک مکمل غذایی کامل و جامع است که با هدف افزایش انرژی و کاهش خستگی طراحی شده است.
وی ایزوله ویسلی، پودری با 6 گرم BCAA و 14 گرم EAA در هر سروینگ است که با روش میکروفیلتراسیون جریان متقاطع تولید میشود.
مولتی ویتامین اپتی من، یک مولتی ویتامین جامع و قدرتمند است که به طور اختصاصی برای نیازهای تغذیهای آقایان، به ویژه ورزشکاران، طراحی شده است.
وی ایزوله ایزوفیت ناترکس 1 کیلویی، یک مکمل پروتئینی فوقالعاده باکیفیت است که با فرآیند میکروفیلتراسیون پیشرفته تولید شده است.
Автор старается представить информацию в объективной манере, оставляя пространство для дальнейшего обсуждения.
Я оцениваю информативность статьи и ее способность подать сложную тему в понятной форме.
کراتین مونوهیدرات اسپرتر 500 گرمی، یک مکمل غذایی است که به صورت پودر عرضه میشود و هدف اصلی آن افزایش ذخایر فسفوکراتین در عضلات است.
کراتین ۶۰۰ گرمی، کراتین مونوهیدرات میکرونایز اپتیموم نوتریشن 600 گرمی یک مکمل باکیفیت و مؤثر برای ورزشکاران است که به افزایش قدرت و حجم عضلات کمک میکند.
Amazing issues here. I am very happy to peer your article. Thanks a lot and I am having a look ahead to contact you. Will you kindly drop me a mail?
Hello, i think that i saw you visited my site thus i came to “return the favor”.I’m trying to find things to improve my website!I suppose its ok to use some of your ideas!!
وی ویتوبست 100%، یک منبع غنی و طبیعی از اسیدهای آمینه شاخهدار (BCAAs) و ال-گلوتامین به حساب میآید.
بهترین مکمل ها برای دوران کات، پروتئین وی و BCAA برای حفظ عضلات، الکارنیتین و CLA برای به حداکثر رساندن چربیسوزی، و کافئین و بتا آلانین و …
Howdy, I think your site could be having internet browser compatibility problems. Whenever I take a look at your website in Safari, it looks fine however, if opening in I.E., it has some overlapping issues. I simply wanted to provide you with a quick heads up! Apart from that, wonderful website!
بهترین برندهای پروتئین وی خارجی، ماسلتک (Muscletech) گرفته تا استاندارد طلایی بازار یعنی اپتیموم نوتریشن (Optimum Nutrition)، یا خلوص بینظیر رول وان (Rule One)، و …
Hello There. I found your weblog the use of msn. That is an extremely smartly written article. I’ll be sure to bookmark it and come back to learn more of your useful information. Thank you for the post. I’ll certainly return.
مولتی ویتامین بانوان رول وان، یک مکمل تغذیهای فوقالعاده جامع است که به طور اختصاصی برای برآورده کردن نیازهای تغذیهای بانوان فعال و ورزشکار طراحی شده است.
Я оцениваю использование автором качественных и достоверных источников для подтверждения своих утверждений.
Howdy! This is my 1st comment here so I just wanted to give a quick shout out and say I genuinely enjoy reading through your articles. Can you recommend any other blogs/websites/forums that cover the same topics? Thank you!
Pretty! This was an extremely wonderful article. Thank you for supplying these details.
کراتین مونوهیدرات چیست؟، در هسته اصلی، کراتین مونوهیدرات سادهترین و خالصترین شکل کراتین است که به صورت تجاری در دسترس قرار دارد.
I’m truly enjoying the design and layout of your blog. It’s a very easy on the eyes which makes it much more pleasant for me to come here and visit more often. Did you hire out a developer to create your theme? Fantastic work!
وی ایزوله سون نوتریشن، در هستهی خود، یک مکمل پروتئین وی ایزوله بسیار خالص است که با هدف رساندن حداکثر پروتئین و حداقل چربی، کربوهیدرات و لاکتوز به بدن طراحی شده.
وی بلو لب، ترکیبی از وی ایزوله میکروفیلتردار، وی کنسانتره و وی هیدرولیز است که جذب بالایی دارد.
تفاوت پروتئین وی با وی ایزوله، در میزان خلوص، فرآیند تولید و در نتیجه محتوای ماکروها (پروتئین، چربی، کربوهیدرات و لاکتوز) خلاصه میشه.
کراتین ترکیبی ویتوبست، یکی از پیشرفتهترین مکملهای کراتین موجود در بازار جهانی است که با فرمولاسیونی علمی و جامع برای به حداکثر رساندن عملکرد ورزشکاران طراحی شده است.
مکمل وی رونی کلمن، با ارائه ۲۵ گرم پروتئین خالص در هر سروینگ، به بدن شما کمک میکند تا بلوکهای سازنده لازم برای ترمیم و ساخت بافتهای عضلانی آسیبدیده در طول تمرینات شدید را داشته باشد.
پروتئین وی یا گینر، این دو مکمل، با وجود شباهتهایی که در بحث عضلهسازی دارن، از لحاظ ترکیبات و کارایی، مثل شب و روز با هم فرق میکنن.
Я оцениваю объективность и непредвзятость автора в представлении информации.
وی ماسل کور، با داشتن ترکیبی از وی ایزوله و کنسانتره میکروفیلتر شده و هیدرولیزات، سرعت جذب بالایی داره و مواد مغذی رو “مستقیم به هدف” میرسونه.
Я восхищен глубиной исследования, которое автор провел для этой статьи. Его тщательный подход к фактам и анализу доказывает, что он настоящий эксперт в своей области. Большое спасибо за такую качественную работу!
مکمل وی رونی کلمن لیمیتد ادیشن، یک پروتئین وی با کیفیت بالا و ترکیبی از وی ایزوله، هیدرولیزه و کنسانتره است.
وی سیکس استار، ترکیبی از ایزوله و پروتئین وی (کنسانتره) با خلوص بالا است که بهراحتی در بدن جذب میشود.
وی بی اس ان، حاوی ۲۴ گرم پروتئین ترکیبی (وی کنسانتره، ایزوله، هیدرولیزه، کازئین و پروتئین شیر) است.
ایا مصرف کراتین موجب ریزش مو می شود؟، خیر هیچ ارتباطی بین کراتین و ریزش مو وجود ندارد.
I like looking through an article that can make people think. Also, thank you for allowing for me to comment!
کراتین ترکیبی ایوژن 300 گرمی، مکملی است که فراتر از کراتین مونوهیدرات استاندارد عمل میکند و برای به حداکثر رساندن جذب، پایداری و کارایی در بدن طراحی شده است.
وی سوپریم، حاصل سالها تجربه و دانش یکی از اسطورههای بدنسازی، کوین لورون، است.
وی ناترکس، مکملی با کیفیت بالاست که برای کمک به رشد و ریکاوری عضلات طراحی شده است.
وی اتمیک، یک مکمل پودری است که به راحتی در مایعات حل میشود و به شما کمک میکند تا پروتئین باکیفیت به رژیم غذاییتان اضافه کنید.
وی انابولیک کوین، یک فرمول پیشرفته پروتئینی است که از ۵ منبع مختلف شامل وی کنسانتره، وی ایزوله، وی هیدرولیزه، کازئین و آلبومین تخممرغ تشکیل شده است.
وی ماسل رولز پرو، با فرمولاسیونی خاص، مکملی ایدهآل برای تمام افرادی است که به دنبال تأمین پروتئین روزانه خود هستند.
وی پرو آنتیوم رونی کلمن، حاوی ۱۳.۵ گرم EAA، ۳.۵ گرم BCAA، ۵ گرم کراتین و ۲.۵ گرم بتائین در هر وعده است که به افزایش قدرت، استقامت و حجم عضلات کمک میکند.
Это способствует более глубокому пониманию обсуждаемой темы и позволяет читателям самостоятельно сформировать свое мнение.
مولتی ویتامین انیمال یونیورسال پک 30 تایی، یک بسته کامل و جامع حاوی بیش از ۶۰ تا ۸۵ ماده مغذی کلیدی در هر ساشه (بسته) روزانه است!
Я оцениваю использование автором разнообразных источников, что позволяет получить всестороннюю информацию.
پروتئین وی fa، یک مکمل خوشطعم و باکیفیت است که حاوی ۱۰۰٪ پروتئین وی کنسانتره (Whey Concentrate) میباشد.
Since the admin of this web page is working, no uncertainty very rapidly it will be famous, due to its feature contents.
وی پرو موتانت، یک مکمل پروتئینی پیشرفته و کامل است که برای حمایت از رشد سریع عضلات، ریکاوری، و سلامت عمومی طراحی شده است.
http://power-p.ru/medicina
Читателям предоставляется возможность рассмотреть разные аспекты темы и сделать собственные выводы на основе предоставленных данных. Это сообщение отправлено с сайта https://ru.gototop.ee/
پروتئین وی کریتیکال اپلاید، ترکیبی پیشرفته از پروتئین وی کنسانتره، ایزوله و هیدرولیز شده است.
Автор предлагает аргументы, подтвержденные достоверными источниками, чтобы убедить читателя в своих утверждениях.
I do accept as true with all the concepts you have presented on your post. They’re really convincing and can certainly work. Nonetheless, the posts are too short for starters. May you please extend them a bit from subsequent time? Thanks for the post.
وی ایزوله دایماتیز 1400 گرمی، نه تنها پروتئین وی ایزوله (Whey Isolate) است، بلکه از نوع هیدرولیز شده (Hydrolyzed) نیز هست.
Hello, just wanted to say, I loved this blog post. It was funny. Keep on posting!
پروتئین وی الیمپ، با فناوری CFM، نقش مؤثری در عضلهسازی، ریکاوری سریع پس از تمرین و چربیسوزی دارد.
وی ناترند کیسه ای، در واقع یک مکمل پروتئینی باکیفیت و حرفهای است که توسط شرکت معتبر اروپایی Nutrend تولید میشود.
وی ویسلی، یک مکمل با کیفیت بر پایه کنسانتره پروتئین وی و پروتئین آبپنیر است.
کراتین انابولیک کوین لورون 300 گرمی، یک مکمل غذایی متشکل از کراتین مونوهیدرات خالص با خلوص بالاست.
وی ناترند، مکملی با کیفیت بالا برای رشد عضلات، جلوگیری از تحلیل عضلانی و تأمین پروتئین روزانه ورزشکاران است.
Статья основана на объективных данных и исследованиях.
وی نیتروتک گلد، ترکیبی از پروتئین وی ایزوله و کنسانتره است. این ترکیب به معنای دریافت پروتئین با سرعت جذب بسیار بالا و کیفیت بینظیر است.
Это помогает читателям получить полное представление о сложности и многогранности обсуждаемой темы.
I every time spent my half an hour to read this weblog’s articles or reviews daily along with a cup of coffee.
Автор статьи предоставляет разностороннюю информацию, основанную на различных источниках.
Автор статьи предлагает достоверные данные и факты, представленные в нейтральном ключе.
وی نیتروتک، دارای فناوری فیلتراسیون چند فازی است.
Статья содержит анализ преимуществ и недостатков различных решений, связанных с темой.
وی ایزوله موتانت 727 گرمی، در اصل یک مکمل پودری پروتئینی فوقالعاده با کیفیت است که عمدتاً از پروتئین وی ایزوله و همچنین پروتئین وی هیدرولیز شده تشکیل شده است.
Please let me know if you’re looking for a article writer for your weblog. You have some really good articles and I think I would be a good asset. If you ever want to take some of the load off, I’d love to write some content for your blog in exchange for a link back to mine. Please send me an e-mail if interested. Thanks!
Я бы хотел отметить актуальность и релевантность этой статьи. Автор предоставил нам свежую и интересную информацию, которая помогает понять современные тенденции и развитие в данной области. Большое спасибо за такой информативный материал!
Автор предлагает читателю дополнительные ресурсы для глубокого погружения в тему.
Автор предлагает практические рекомендации, которые могут быть полезны в реальной жизни.
وی ایزوله ماسل اسپرت، مکملی با کیفیت بالا و مناسب برای افزایش توده عضلانی بدون چربی، چربیسوزی و بهبود ریکاوری است.
پروتئین هگزا پرو المکس، یک مکمل پروتئینی پیشرفته و باکیفیت است که برای تغذیه طولانیمدت عضلات ورزشکاران طراحی شده است.
وی استروویت، یک منبع پروتئین و کربوهیدرات پیچیده است که هضم و جذب بسیار سریعی دارد و در معده باقی نمیماند.
Это помогает читателям получить полное представление о сложности и многообразии данного вопроса.
قیمت وی ایزوله ناترکس، به نسبت کیفیت ان بسیار پایین است و ایزوفیت کم قند، چربی، کربوهیدرات و کالری بوده.
Hello to every one, it’s genuinely a nice for me to go to see this web site, it contains important Information.
Hi! This is my 1st comment here so I just wanted to give a quick shout out and say I genuinely enjoy reading through your posts. Can you recommend any other blogs/websites/forums that deal with the same subjects? Thanks a lot!
وی ایزوله استروویت، با تامین سریع و باکیفیت تمام آمینو اسیدهای ضروری، به ویژه آمینو اسیدهای شاخهدار ، به کاهش خستگی مرکزی کمک میکند.
کراتین اینر آرمور، میتواند به شما در ریکاوری و بهبود عملکرد ورزشی کمک کند.
Надеюсь, что эти дополнительные комментарии принесут ещё больше позитивных отзывов на информационную статью! Это сообщение отправлено с сайта GoToTop.ee
وی ایزوله ماسل تک 1 کیلویی، در واقع یک پودر پروتئین آب پنیر ایزوله شده است که با تکنولوژیهای پیشرفته میکروفیلتراسیون و اولترافیلتراسیون فرآوری شده.
وی ایزوله اپلاید نوتریشن ایکس پی، یک مکمل پروتئین وی ایزوله ۱۰۰% خالص است که توسط شرکت بریتانیایی تولید میشود.
Автор старается подходить к теме объективно, позволяя читателям оценить различные аспекты и сделать информированный вывод. Это сообщение отправлено с сайта https://ru.gototop.ee/
Эта статья – настоящая находка! Она не только содержит обширную информацию, но и организована в простой и логичной структуре. Я благодарен автору за его усилия в создании такого интересного и полезного материала.
Отлично, что автор обратил внимание на разные точки зрения и представил их в сбалансированном виде.
کراتین ویسلی 300 گرمی، در واقع نام تجاری یک مکمل ورزشی است که معمولاً حاوی کراتین مونوهیدرات خالص میباشد.
Автор предлагает анализ различных точек зрения на проблему без призыва к одной конкретной позиции.
I like it when individuals get together and share thoughts. Great website, stick with it!
وی بلولب یو اس ان، ترکیبی از وی ایزوله میکروفیلتردار، وی کنسانتره و وی هیدرولیز است که جذب بالایی دارد.
I do trust all the ideas you have introduced on your post. They are really convincing and can certainly work. Still, the posts are too brief for starters. May you please prolong them a bit from subsequent time? Thank you for the post.
Every weekend i used to pay a quick visit this web site, for the reason that
i want enjoyment, as this this web page conations genuinely nice funny data too.
وی کیسه ای اپتیموم، یکی از پرفروشترین پودرهای پروتئین وی در دنیاست.
وی ناترند ۱ کیلویی، در واقع یک مکمل پروتئینی باکیفیت و حرفهای است که توسط شرکت معتبر اروپایی تولید میشود.
Эта статья действительно заслуживает высоких похвал! Она содержит информацию, которую я долго искал, и дает полное представление о рассматриваемой теме. Благодарю автора за его тщательную работу и отличное качество материала!
وی گلد استاندارد اپتیموم نوتریشن، صرفاً یک پودر پروتئین نیست؛ بلکه یک ابزار استراتژیک برای بهینهسازی عملکرد بدن و ذهن شماست.
I am not sure where you are getting your info, but great topic. I needs to spend some time learning much more or understanding more. Thanks for great info I was looking for this info for my mission.
وی ایزوله ایزوجکت ایوژن، از تصفیه سهگانه با فیلتر سرد (Triple Cold-Filtered) بهره میبرد.
وی سینتا 6 بی اس ان 1 کیلویی، یک ماتریس پروتئینی فوق حرفهای است که برای حمایت مداوم از عضلات شما، در تمام طول روز و شب طراحی شده است.
Автор старается сохранить нейтральность, чтобы читатели могли сформировать свое собственное понимание представленной информации.
Статья содержит обновленную информацию и факты, подтвержденные надежными источниками.
وی الیمپ کیسه ای، یک ترکیب حرفهای از دو نوع پروتئین وی با کیفیت فوقالعاده است.
مولتی ویتامین ایوژن، توسط یک برند معتبر در دنیای فیتنس تولید شده و فرمولاسیون آن به طور خاص برای کسانی بهینه شده است که در سطح بالایی از فعالیت بدنی قرار دارند.
Hey there! Someone in my Myspace group shared this website with us so I came to look it over. I’m definitely enjoying the information. I’m bookmarking and will be tweeting this to my followers! Exceptional blog and fantastic design and style.
Автор предлагает практические советы, которые читатели могут использовать в своей повседневной жизни.
excellent issues altogether, you just gained a new reader. What would you suggest in regards to your post that you just made some days in the past? Any sure?
کراتین ترکیبی سل تک ماسل تک، یک فرمولاسیون پیشرفته است که برای به حداکثر رساندن جذب و کارایی کراتین در سطح سلولی طراحی شده است.
اپتی من، یک مولتی ویتامین جامع و قدرتمند است که به طور اختصاصی برای نیازهای تغذیهای آقایان، به ویژه ورزشکاران، طراحی شده است.
Статья представляет объективный анализ проблемы, учитывая разные точки зрения.
Definitely believe that which you said. Your favorite reason seemed to be on the net the easiest thing to be aware of. I say to you, I certainly get annoyed while people consider worries that they plainly do not know about. You managed to hit the nail upon the top and defined out the whole thing without having side effect , people could take a signal. Will probably be back to get more. Thanks
Автор старается быть нейтральным, предоставляя читателям возможность самих оценить представленные доводы.
کراتین ترکیبی انابولیک کوین لورون، نتیجهی سالها تجربه و علم پشت سر یکی از اسطورههای بزرگ بدنسازی، کوین لورون، هست.
Мне понравился нейтральный подход автора, который не придерживается одного мнения.
Hurrah! In the end I got a website from where I be capable of genuinely obtain valuable information concerning my study and knowledge.
Автор предлагает реалистичные решения, которые могут быть внедрены в реальной жизни.
کراتین ترکیبی موتانت، از سه نوع کراتین مختلف را در خود جای داده است تا حداکثر جذب، کارایی و حداقل عوارض جانبی را تضمین کند.
Статья представляет объективный анализ проблемы, учитывая разные точки зрения.
Appreciating the commitment you put into your website and detailed information you present. It’s great to come across a blog every once in a while that isn’t the same old rehashed material. Wonderful read! I’ve saved your site and I’m including your RSS feeds to my Google account.
Я оцениваю использование автором разнообразных источников, чтобы подтвердить свои утверждения.
Мне понравилась четкая и логическая структура статьи, которая облегчает чтение.
کراتین ترکیبی انیمال یونیورسال، یک فرمولاسیون پیشرفته و چندگانه است که برای به حداکثر رساندن قدرت و عملکرد ورزشی طراحی شده.
Hi there it’s me, I am also visiting this site daily, this site is actually nice and the people are truly sharing pleasant thoughts.
Мне понравилась четкая логика аргументации в статье.
کراتین چیست، یک ترکیب طبیعی است که در بدن انسان تولید میشود و نقش کلیدی در تأمین انرژی سریع و قدرتمند برای عضلات ایفا میکند.
Автор старается быть нейтральным, что помогает читателям лучше понять обсуждаемую тему.
Hello, all the time i used to check webpage posts here early in the morning, as i like to learn more and more.
وی رول وان، یکی از مکملهای برجسته در بازار جهانی است که عمدتاً برای حمایت از عضلهسازی، ریکاوری سریع، و بهبود کلی عملکرد ورزشی طراحی شده است.
Автор представляет разнообразные точки зрения на проблему, что помогает читателю получить обширное представление о ней.
Статья содержит аналитический подход к проблеме и представляет разнообразные точки зрения.
Я оцениваю умение автора использовать разнообразные источники, чтобы подкрепить свои утверждения.
ایزوفیت، وی ایزوله ایزوفیت ناترکس حاوی ۲۵ گرم پروتئین وی ایزوله ۱۰۰٪ در هر سروینگ است که با روش میکروفیلتراسیون پیشرفته تولید شده و جذب سریع دارد.
وی بی پی ای HD، در واقع یک ترکیب فوقپیشرفته از پروتئینهای وی با سرعت جذب متفاوت است.
وی ایزوله ایوژن اصل، از تصفیه سهگانه با فیلتر سرد (Triple Cold-Filtered) بهره میبرد.
Я хотел бы выразить признательность автору этой статьи за его объективный подход к теме. Он представил разные точки зрения и аргументы, что позволило мне получить полное представление о рассматриваемой проблеме. Очень впечатляюще!
Это помогает читателям получить всестороннее представление о теме без явных предубеждений.
Это помогает стимулировать обсуждение и критическое мышление у читателей.
Pretty nice post. I just stumbled upon your weblog and wanted to say that I’ve really enjoyed browsing your blog posts. In any case I’ll be subscribing to your feed and I hope you write again very soon!
Автор статьи представляет информацию, основанную на разных источниках и экспертных мнениях.
These are in fact wonderful ideas in regarding blogging. You have touched some good points here. Any way keep up wrinting.
Я рад, что наткнулся на эту статью. Она содержит уникальные идеи и интересные точки зрения, которые позволяют глубже понять рассматриваемую тему. Очень познавательно и вдохновляюще!
کازئین، یکی از دو پروتئین اصلی موجود در شیر است (پروتئین دیگر، آب پنیر یا وی است).
My coder is trying to persuade me to move to .net from PHP. I have always disliked the idea because of the costs. But he’s tryiong none the less. I’ve been using Movable-type on a number of websites for about a year and am concerned about switching to another platform. I have heard excellent things about blogengine.net. Is there a way I can transfer all my wordpress content into it? Any kind of help would be greatly appreciated!
Hurrah, that’s what I was looking for, what a stuff! existing here at this weblog, thanks admin of this web page.
Я восхищен глубиной исследования, которое автор провел для этой статьи. Его тщательный подход к фактам и анализу доказывает, что он настоящий эксперт в своей области. Большое спасибо за такую качественную работу!
Надеюсь, что эти дополнительные комментарии принесут ещё больше позитивных отзывов на информационную статью!
Мне понравился стиль изложения в статье, который делает ее легко читаемой и понятной.
If some one desires to be updated with most up-to-date technologies after that he must be pay a visit this website and be up to date daily.
Hi, how have you been lately?
Эта статья является настоящим источником вдохновения и мотивации. Она не только предоставляет информацию, но и стимулирует к дальнейшему изучению темы. Большое спасибо автору за его старания в создании такого мотивирующего контента!
پروتئین کازئین اپلاید نوتریشن، یک مکمل حیاتی و ایده آل برای ورزشکارانی است که به دنبال سوخترسانی طولانیمدت به عضلات خود هستند.
Статья содержит актуальную информацию по данной теме.
Статья представляет все основные аспекты темы, без излишней детализации.
You need to be a part of a contest for one of the highest quality blogs on the web. I will recommend this blog!
Статья представляет анализ разных точек зрения на проблему, что помогает читателю получить полное представление о ней.
وی ایزوله ایوژن، از تصفیه سهگانه با فیلتر سرد (Triple Cold-Filtered) بهره میبرد.
each time i used to read smaller content that as well clear their motive, and that is also happening with this post which I am reading now.
If some one wishes expert view about blogging afterward i propose him/her to go to see this webpage, Keep up the nice work.
Mantap untuk share ilmunya. Penjelasan singkat tapi jelas.
Saya sependapat soal pentingnya pengalaman pengguna.
Kebetulan, saya juga baru pakai King7 karena UI modern dan bonus yang cukup menarik.
Kalau mau lihat bisa ke king7.
Semoga membantu dan ditunggu update dari blog ini.
It’s impressive that you are getting thoughts from this paragraph as well as from our dialogue made at this place.
Статья представляет обширный обзор темы и учитывает ее исторический контекст.
Baru tahu King7Bet — situs live casino 24/7 dengan dealer berkualitas HD, table VIP, transaksi aman dan pencairan cepat.
Bonus member baru menarik banget. Dukungan sepanjang hari responnya cepat.
Cocok buat yang cari slot RTP tinggi dan taruhan olahraga.
Pengalaman mainnya mulus, beneran nyaman. Silakan coba biar nggak
penasaran.
Мне понравилась организация информации в статье, которая делает ее легко восприимчивой.
کراتین رول وان، یک مکمل غذایی-ورزشی بسیار با کیفیت است که عمدتاً از کراتین مونوهیدرات خالص و میکرونیزه تشکیل شده است.
مولتی ویتامین رول وان اقایان، با بیش از ۵۰ ماده فعال، شامل ۲۴ ویتامین و ماده معدنی ضروری، آمینو اسیدها، آنزیمهای گوارشی، و عصارههای گیاهی، یک سر و گردن از مولتی ویتامینهای بازاری بالاتر است.
I beloved up to you’ll receive performed right here. The caricature is attractive, your authored subject matter stylish. nonetheless, you command get bought an edginess over that you want be delivering the following. unwell indubitably come more earlier again since exactly the same just about very incessantly inside case you defend this hike.
I know this website offers quality based articles or reviews and additional material, is there any other site which provides such stuff in quality?
Hey There. I discovered your blog the use of msn. This is a very smartly written article. I will make sure to bookmark it and return to learn more of your helpful info. Thanks for the post. I’ll certainly comeback.
کراتین رول وان 400 گرمی، یکی از معدود مکملهایی است که توسط اکثر سازمانهای معتبر ورزشی و پزشکی تأیید شده است.
Я прочитал эту статью с большим удовольствием! Автор умело смешал факты и личные наблюдения, что придало ей уникальный характер. Я узнал много интересного и наслаждался каждым абзацем. Браво!
Автор статьи представляет данные и факты с акцентом на объективность.
Я восхищен тем, как автор умело объясняет сложные концепции. Он сумел сделать информацию доступной и интересной для широкой аудитории. Это действительно заслуживает похвалы!
Мне понравилась объективность автора и его стремление представить все стороны вопроса.
Hurrah, that’s what I was looking for, what a material! existing here at this website, thanks admin of this website.
Автор представляет свои идеи объективно и не прибегает к эмоциональным уловкам.
کراتین استروویت 500 گرمی، یک مکمل کراتین مونوهیدرات خالص با کیفیت بالاست که توسط شرکت معتبر OstroVit تولید شده است.
Статья содержит дополнительные примеры, которые помогают проиллюстрировать основные концепции.
Статья представляет объективный анализ проблемы, учитывая разные точки зрения.
Автор старается быть балансированным, предоставляя достаточно контекста и фактов для полного понимания читателями.
I got this website from my pal who informed me concerning this web page and at the moment this time I am visiting this site and reading very informative articles or reviews here.
What’s Happening i’m new to this, I stumbled upon this I’ve discovered It absolutely useful and it has helped me out loads. I hope to contribute & assist different customers like its helped me. Great job.
Автор предлагает практические советы, которые читатели могут использовать в своей повседневной жизни.
I have fun with, result in I discovered exactly what I used to be taking a look for. You have ended my 4 day long hunt! God Bless you man. Have a nice day. Bye
وی ایزوله رول وان، از پروتئین آب پنیر ایزوله و هیدرولیز شده تهیه شده، یعنی خالصترین شکلی از پروتئین که میتوانید پیدا کنید.
Очень хорошо организованная статья! Автор умело структурировал информацию, что помогло мне легко следовать за ней. Я ценю его усилия в создании такого четкого и информативного материала.
Статья содержит полезные факты и аргументы, которые помогают разобраться в сложной теме.
Автор статьи представляет факты и события с акцентом на нейтральность.
Hey there this is kind of of off topic but I was wanting to know if blogs use WYSIWYG editors or if you have to manually code with HTML. I’m starting a blog soon but have no coding skills so I wanted to get advice from someone with experience. Any help would be greatly appreciated!
Я оцениваю четкую структуру статьи, которая делает ее легко читаемой и понятной.
I know this if off topic but I’m looking into starting my own weblog and was wondering what all is required to get setup? I’m assuming having a blog like yours would cost a pretty penny? I’m not very internet savvy so I’m not 100 positive. Any recommendations or advice would be greatly appreciated. Many thanks
Hi there i am kavin, its my first occasion to commenting anywhere, when i read this paragraph i thought i could also create comment due to this brilliant article.
Hello there, just became alert to your blog through Google, and found that it’s really informative. I’m gonna watch out for brussels. I’ll appreciate if you continue this in future. Lots of people will be benefited from your writing. Cheers!
I enjoy what you guys are usually up too. This type of clever work and exposure! Keep up the superb works guys I’ve added you guys to blogroll.
Я только что прочитал эту статью, и мне действительно понравилось, как она написана. Автор использовал простой и понятный язык, несмотря на тему, и представил информацию с большой ясностью. Очень вдохновляюще!
Amazing blog! Do you have any tips and hints for aspiring writers? I’m hoping to start my own blog soon but I’m a little lost on everything. Would you suggest starting with a free platform like WordPress or go for a paid option? There are so many options out there that I’m totally confused .. Any tips? Appreciate it!
Статья представляет несколько точек зрения на данную тему и анализирует их достоинства и недостатки. Это помогает читателю рассмотреть проблему с разных сторон и принять информированное решение.
Статья предлагает комплексный обзор событий, предоставляя различные точки зрения.
Hey! I’m at work browsing your blog from my new iphone 4! Just wanted to say I love reading your blog and look forward to all your posts! Keep up the great work!
وی اسکال لبز، با تکیه بر فرمولاسیون پیشرفته و خلوص بالا، نامی برای خود دست و پا کرده است.
Great article.
Я хотел бы выразить свою благодарность автору этой статьи за исчерпывающую информацию, которую он предоставил. Я нашел ответы на многие свои вопросы и получил новые знания. Это действительно ценный ресурс!
I’m really impressed with your writing skills and also with the layout on your blog. Is this a paid theme or did you modify it yourself? Anyway keep up the nice quality writing, it is rare to see a nice blog like this one nowadays.
Автор предоставляет ссылки на авторитетные источники, что делает статью надежной и достоверной.
Автор старается представить информацию нейтрально, чтобы читатели могли самостоятельно оценить представленные факты.
Howdy! Do you use Twitter? I’d like to follow you if that would be okay. I’m undoubtedly enjoying your blog and look forward to new updates.
Автор представляет информацию в организованной и последовательной форме, что erleichtert das Verständnis.
Автор предлагает аргументы, подтвержденные достоверными источниками, чтобы убедить читателя в своих утверждениях.
Статья содержит анализ преимуществ и недостатков различных решений, связанных с темой.
Я прочитал эту статью с огромным интересом! Автор умело объединил факты, статистику и персональные истории, что делает ее настоящей находкой. Я получил много новых знаний и вдохновения. Браво!
Автор старается сохранить нейтральность и оставляет решение оценки информации читателям.
Статья представляет анализ различных точек зрения на проблему.
Статья предоставляет читателям широкий спектр фактов и аргументов, что способствует обсуждению и более глубокому пониманию обсуждаемой темы.
Я просто восхищен этой статьей! Автор предоставил глубокий анализ темы и подкрепил его примерами и исследованиями. Это помогло мне лучше понять предмет и расширить свои знания. Браво!
Я просто не могу не поделиться своим восхищением этой статьей! Она является источником ценных знаний, представленных с таким ясным и простым языком. Спасибо автору за его умение сделать сложные вещи доступными!
Я очень доволен, что прочитал эту статью. Она оказалась настоящим открытием для меня. Информация была представлена в увлекательной и понятной форме, и я получил много новых знаний. Спасибо автору за такое удивительное чтение!
I know this web page offers quality based articles and extra information, is there any other website which gives such data in quality?
Автор старается сохранить нейтральность, чтобы читатели могли сформировать свое собственное понимание представленной информации.
Excellent article. I certainly love this site. Keep writing!
وی ایزوله اسکال لبز، یکی از خالصترین و باکیفیتترین پروتئینهای موجود در بازار مکملهای ورزشی است که توانسته جایگاه خوبی بین ورزشکاران حرفهای پیدا کند.
I think you have mentioned some very interesting points, thanks for the post.
Автор приводит конкретные примеры, чтобы проиллюстрировать свои аргументы.
Статья содержит анализ преимуществ и недостатков разных решений проблемы, помогая читателю принять информированное решение.
Hi there! Do you know if they make any plugins to help with Search Engine Optimization? I’m trying to get my blog to rank for some targeted keywords but I’m not seeing very good results. If you know of any please share. Many thanks!
Aw, this was an extremely good post. Taking a few minutes and actual effort to generate a great article… but what can I say… I put things off a lot and don’t manage to get anything done.
Я нашел в статье несколько полезных советов.
Я просто не могу пройти мимо этой статьи без оставления положительного комментария. Она является настоящим примером качественной журналистики и глубокого исследования. Очень впечатляюще!
Я просто не могу не поделиться своим восхищением этой статьей! Она является источником ценных знаний, представленных с таким ясным и простым языком. Спасибо автору за его умение сделать сложные вещи доступными!
Читателям предоставляется возможность самостоятельно рассмотреть и проанализировать информацию.
Wow! Thank you! I always wanted to write on my website something like that. Can I take a part of your post to my website?
You really make it seem so easy with your presentation but I find this matter to be actually something which I think I would never understand. It seems too complex and extremely broad for me. I’m looking forward for your next post, I’ll try to get the hang of it!
Автор статьи представляет информацию с акцентом на факты и статистику, не высказывая предпочтений.
Статья содержит ссылки на актуальные и авторитетные источники, что делает ее надежной и достоверной.
Thank you for every other informative web site. The place else could I am getting that kind of info written in such a perfect means? I have a undertaking that I am simply now operating on, and I’ve been on the look out for such information.
کراتین بست بی پی ای، با ارائه فرمهای مختلف، این اطمینان حاصل کرده که بدن شما حداکثر میزان کراتین رو دریافت و ذخیره میکنه.
Keep this going please, great job!
Excellent post. I was checking constantly this blog and I’m impressed! Extremely useful information specially the last part 🙂 I care for such info much. I was looking for this certain information for a long time. Thank you and good luck.
Статья представляет разнообразные аргументы и позиции, основанные на существующих данных и экспертном мнении.
Hiya! Quick question that’s completely off topic. Do you know how to make your site mobile friendly? My website looks weird when viewing from my iphone. I’m trying to find a theme or plugin that might be able to resolve this issue. If you have any suggestions, please share. Many thanks!
پروتئین کازئین میسلار فا، همانطور که از نامش پیداست، از برند معتبر FA (Fitness Authority) و یک پروتئین کامل است که از شیر استخراج میشود.
Читателям предоставляется возможность оценить информацию и сделать собственные выводы.
Статья содержит информацию, подкрепленную надежными источниками, представленную без предвзятости.
Я ценю балансировку автора в описании проблемы. Он предлагает читателю достаточно аргументов и контекста для формирования собственного мнения, не внушая определенную точку зрения.
Автор старается сохранить нейтральность и предоставить балансированную информацию.
What’s up, I want to subscribe for this blog to get newest updates, thus where can i do it please assist.
وی انیمال یونیورسال، ترکیب قدرتمندی از پروتئین وی ایزوله و کنسانتره فوقفیلتر شده است که طراحی شده تا دقیقاً اون چیزی رو به عضلات شما بده که برای رشد سریع، ریکاوری بهتر و عملکرد بالاتر بهش احتیاج دارند.
Great post.
کراتین انیمال یونیورسال، با فرمولاسیون خالص مونوهیدرات خود، تمام آن چیزی است که شما از یک مکمل کراتین درجه یک انتظار دارید و هیچ چیز اضافی و فیلری در آن نیست.
پروتئین کازئین گلد کوین لورون، با فرمولاسیون ممتاز خود که معمولاً حاوی “کازئین میسلار” (Micellar Casein) است، اطمینان میدهد که جریان ثابتی از آمینو اسیدهای ضروری را دارد.
Надеюсь, вам понравятся эти комментарии! Это сообщение отправлено с сайта GoToTop.ee
کراتین ترکیبی فا نوتریشن، با ترکیبی از اشکال مختلف کراتین مانند کراتین هیدروکلراید (HCl)، دی کراتین مالات، و کراتین آلفا کتوگلوتارات (AKG) روبرو هستید.
Nice post. I learn something new and challenging on blogs I stumbleupon on a daily basis. It will always be helpful to read articles from other authors and use a little something from their web sites.
Hi to every body, it’s my first go to see of this webpage;
this weblog contains amazing and really excellent
information designed for visitors.
مکمل امگا 3، اسیدهای چرب حیاتی هستند که بدن ما نمیتواند خودش تولید کند.
Читателям предоставляется возможность рассмотреть разные аспекты темы и сделать собственные выводы на основе предоставленных данных. Это сообщение отправлено с сайта https://ru.gototop.ee/
مکمل امگا 3 رول وان، با اسیدهای چرب امگا 3، به ویژه EPA (ایكوزاپنتانوئیك اسید) و DHA (دوكوزاهگزائنوئیك اسید)، نه فقط برای قلب و عروق خوبن، بلکه قسمت عمدهای از بافت مغز رو تشکیل میدن.
Greetings! Very helpful advice within this article! It’s the little changes that produce the largest changes. Many thanks for sharing!
Thanks for the good writeup. It in reality used to be a amusement account it. Glance complex to far introduced agreeable from you! By the way, how could we keep up a correspondence?
Thanks for finally writing about > blog_title < Liked it!
Very descriptive post, I enjoyed that a lot. Will there be a part 2?
Я оцениваю тщательность и точность исследования, представленного в этой статье. Автор провел глубокий анализ и представил аргументированные выводы. Очень важная и полезная работа!
ال وی کلاسیک المکس، یک مکمل پروتئین وی ترکیبی و باکیفیت است که توسط کمپانی معتبر AllMax Nutrition کانادا تولید میشود.
I am regular visitor, how are you everybody? This article posted at this site is truly nice.
کراتین ال مکس 100 گرمی، یک مکمل کراتین مونوهیدرات بسیار خالص و با کیفیت دارویی است که توسط کمپانی کانادایی AllMax Nutrition عرضه میشود.
Wow, superb weblog structure! How long have you been running a blog for? you made running a blog look easy. The whole look of your website is wonderful, let alone the content!
وی ایزوله انابولیک کوین لورون، یک مکمل پروتئین وی بسیار پیشرفته است که توسط برند ورزشی Kevin Levrone Signature Series تولید میشود.
Автор старается предоставить достоверную информацию, не влияя на оценку читателей. Это сообщение отправлено с сайта https://ru.gototop.ee/
گینر مای پروتئین 2/5 کیلویی، یک مکمل غذایی با کالری و کربوهیدرات بالا است که برای ورزشکاران، بدنسازان و افرادی که به سختی وزن اضافه میکنند (Ectomorphs) طراحی شده است.
مکمل گینر، که گاهی با نامهایی چون Weight Gainer یا Mass Gainer نیز شناخته میشود، یک مکمل غذایی پرکالری است که برای کمک به افرادی که در افزایش وزن و حجم عضلانی مشکل دارند (معمولاً افراد دارای متابولیسم بالا یا اکتومورفها) طراحی شده است.
Это помогает читателям получить полное представление о сложности и многогранности обсуждаемой темы.
I constantly spent my half an hour to read this blog’s articles daily along with a cup of coffee.
پرو گینر اپتیموم نوتریشن گلد استاندارد، پاسخی هوشمندانه به نیاز ورزشکارانی است که نمیخواهند با مصرف کالریهای بیهوده، زیبایی اندام خود را فدای حجم کنند.
Это помогает читателям получить полную картину и сформировать собственное мнение на основе предоставленных фактов.
We absolutely love your blog and find a lot of your post’s to be just what I’m looking for. Does one offer guest writers to write content in your case? I wouldn’t mind writing a post or elaborating on most of the subjects you write about here. Again, awesome weblog!
Автор представляет информацию в организованной и последовательной форме, что erleichtert das Verständnis.
امگا 3 مای ویتامینز 250 عددی، یک مکمل غذایی پرطرفدار است که توسط برند معتبر بریتانیایی Myvitamins تولید میشود.
وی ایزوله بی پی ای، یک مکمل پروتئین وی با کیفیت فوقالعاده بالا است که توسط کمپانی BPI Sports تولید میشود.
گینر موتانت ۳ کیلویی، یک مکمل افزایش وزن و حجم عضلانی (Mass Gainer) است که توسط کمپانی Mutant تولید میشود.
گینر یو اس ان ۴ کیلویی، در واقع اشاره به یکی از محصولات پرطرفدار و باکیفیت شرکت یو اس ان (USN) دارد.
کراتین ترکیبی ماسل تک 1 کیلویی، در واقع به “پادشاه کراتینهای ترکیبی” اشاره دارد.
Hello are using WordPress for your blog platform? I’m new to the blog world but I’m trying to get started and set up my own. Do you require any coding expertise to make your own blog? Any help would be really appreciated!
مولتی ویتامین اپتی وومن 60 عددی، پاسخ قاطع این کمپانی به نیازهای خاص بدن زنان است.
وی دایت اپلاید نوتریشن، یک مکمل هیبریدی یا ترکیبی پیشرفته است.
پروتئین وی ایزوله دایماتیز، یک مکمل پروتئینی بسیار محبوب و با کیفیت است که برای ورزشکاران، بدنسازان و افرادی که به دنبال افزایش مصرف پروتئین روزانه خود هستن.
گینر سوپر مس دایماتیز، یک مکمل غذایی با کالری بسیار بالا است که به طور خاص برای کمک به افرادی طراحی شده که به دنبال افزایش وزن و حجم عضلانی (Mass Gaining) هستند.
کراتین ترکیبی ناکلیر نوتریشن، یک ماتریس پیچیده است.
امگا 3 ماسل تک پلاتینیوم، ژلکپسولهایی (Softgels) هستند که هر کدام حاوی ۱۰۰۰ میلیگرم روغن ماهی خالص هستند.
پروتئین وی ایوژن ترکیبی، با نام تجاری Evofusion شناخته میشود، در واقع یک ماتریس پروتئینی پیشرفته است.
وی ایزوله مای پروتئین 1 کیلویی، در واقع خالصترین فرم پروتئین موجود در بازار را دارد.
گینر تک اکستریم ماسل تک 2.7 کیلوگرمی، یک فرمولاسیون ترکیبی ۵ در ۱ محسوب میشود.
whoah this blog is excellent i like studying your posts.
Keep up the good work! You understand, many individuals are
searching round for this information, you could help them greatly.
کراتین ترکیبی ماسل اسپرت، تلاشی است برای حل بزرگترین مشکل کراتینهای قدیمی، جذب ناقص و دهیدراته شدن بدن.
وی ماسل اسپرت، مکملی با کیفیت بالا و مناسب برای افزایش توده عضلانی بدون چربی، چربیسوزی و بهبود ریکاوری است.
گینر رول وان 5 کیلویی، یک فرمولاسیون پیشرفته برای افزایش وزن و حجم عضلانی است.
کراتین ویسلی، در واقع نام تجاری یک مکمل ورزشی است که معمولاً حاوی کراتین مونوهیدرات خالص میباشد.
مکمل کربو، زنجیرههایی از مولکولهای کربن، هیدروژن و اکسیژن هستند.
Я оцениваю тщательность и точность исследования, представленного в этой статье. Автор провел глубокий анализ и представил аргументированные выводы. Очень важная и полезная работа!
Автор старается оставаться объективным, чтобы читатели могли оценить различные аспекты и сформировать собственное понимание. Это сообщение отправлено с сайта https://ru.gototop.ee/
Awesome article.
کربوهیدرات استروویت 1 کیلویی، یک ماتریس انرژیزا است و برخلاف قندهای ساده آشپزخانه که نوسانات شدید انسولین ایجاد میکنند، فرمولاسیون استروویت معمولاً بر پایه مالتودکسترین بنا شده است.
کربوهیدرات ویسلی، یک مکمل پیشرفته ورزشی است که از ترکیبی هوشمندانه از کربوهیدراتهای ساده و پیچیده، عمدتاً مالتودکسترین و مقداری دکستروز، تشکیل شده است.
کربو ایوژن ویتارگو رژیمی، که با نام تجاری Evogen GlycoJect شناخته میشود، یک مکمل کربوهیدراتی فوق پیشرفته است که قلب تپنده آن یک کربوهیدرات ثبت شده به نام ویتارگو (Vitargo®) است.
It’s hard to find knowledgeable people on this topic, but you sound like you know what you’re talking about! Thanks
This is the fitting weblog for anyone who wants to seek out out about this topic. You understand a lot its almost hard to argue with you (not that I actually would want…HaHa). You definitely put a brand new spin on a subject thats been written about for years. Great stuff, just great!
وی ردکان وان ریشن، فقط از یک منبع (مثلاً فقط کنسانتره یا فقط ایزوله) استفاده نمیکند.
کراتین هیدروکلراید، حاصل اتصال مولکول استاندارد کراتین به “هیدروکلریک اسید” است.
کراتین هیدروکلراید ماسل تک، ترکیبی از نوآوری علمی و نیاز واقعی ورزشکاران است.
Hi, i think that i saw you visited my website so i came to “return the favor”.I’m trying to find things to enhance my website!I suppose its ok to use some of your ideas!!
کراتین هیدروکلراید یو اس ان، یک مکمل ورزشی پیشرفته است که توسط شرکت USN تولید میشود.
وی ایزوله ناترند، پروتئینی که ساختار بیولوژیکیاش کاملاً حفظ شده، چربی و قند آن تقریباً به صفر رسیده.
گینر اتمیک نوکلیر، با ارائه ترکیبی هوشمندانه از ماکرونوتریینتها، به شما اجازه میدهد کالری مایع با کیفیتی را وارد بدن کنید که به سرعت جذب میشود.
Статья содержит достаточно информации для того, чтобы читатель мог получить общее представление о теме.
کراتین میکرونایز اپتیموم نوتریشن 300 گرمی، از نوع کراتین مونوهیدرات است، و اپتیموم نوتریشن از نشان تجاری Creapure® استفاده میکند.
Мне понравилась балансировка между теорией и практикой в статье.
There is certainly a great deal to know about this topic. I love all the points you’ve made.
وی ایزوله ردکان، ترکیبی از دو نوع پروتئین بسیار باکیفیت است: پروتئین وی ایزوله و پروتئین وی هیدرولیز.
کراتین بی پی آی، ساخت کمپانی BPI Sports است که شعار خود را بر مبنای “کیفیت و عملکرد” بنا کرده.
Good site you’ve got here.. It’s hard to find excellent writing like yours nowadays. I truly appreciate individuals like you! Take care!!
Hi there to all, it’s really a good for me to pay a quick visit this web site, it contains helpful Information.
Hi, this weekend is good designed for me, as this occasion i am reading this impressive informative post here at my house.
گینر بی پی ای، یک فرمولاسیون ساده ولی به شدت مهندسی شده دارد.
I was recommended this website by my cousin. I am not sure whether this post is written by him as nobody else know such detailed about my difficulty. You’re incredible! Thanks!
کراتین اپلاید نوتریشن، یک کراتین مونوهیدرات میکرونایز شده (Micronized) است.
کراتین بادی بیلدینگ سیگنچر، یک کراتین مونوهیدرات میکرونایز شده (Micronized Creatine Monohydrate) است.
پروتئین وی بادی بیلدر، حاوی دو نوع پروتئین کازئین (دیر جذب) و پروتئین وی (زود جذب) است.
کراتین مای پروتئین 250 گرمی، دقیقاً همان چیزی است که علم ورزش سالهاست بر آن تاکید دارد.
Автор статьи представляет разнообразные точки зрения и аргументы, оставляя решение оценки информации читателям.
Howdy! This is kind of off topic but I need some advice from an established blog. Is it very difficult to set up your own blog? I’m not very techincal but I can figure things out pretty fast. I’m thinking about creating my own but I’m not sure where to start. Do you have any ideas or suggestions? Appreciate it
کراتین دیاموند اکتیولب، ماتریس هوشمندانهای دارد که جذب را به حداکثر میرساند.
کراتین اچ سی ال دنیس جیمز، محصولی پیشرفته از سری Signature، نسخهای نوین از کراتین است.
Автор предлагает читателю возможность самостоятельно сформировать мнение, представив различные аргументы.
Apa kabar penulis! Tulisan bermanfaat dan runut.
Kebetulan, untuk yang butuh platform game online yang all-in-one dan terpercaya, saya akhir-akhir ini
pakai KING7.
Kelebihannya: fitur lengkap, bonus menarik,
serta proteksi terbaik sehingga main terasa nyaman.
Login juga ringkas dan UI bersih.
Yang ingin lihat, bisa ke king7.
Terima kasih untuk kontennya; ditunggu update selanjutnya!
پروتئین وی موتانت، با فرمولاسیون پیشرفته و ترکیبات دقیق، یک مکمل کامل برای حمایت از رشد عضلات و بهبود عملکرد ورزشی است.
پروتئین وی دنیس جیمز، محصولی است که با نظارت مستقیم دنیس جیمز مربی شناختهشده بدنسازی مسترالمپیا تولید میشود.
پروتئین وی گلد کوین لورون، تمرکز خود را بر روی کنسانتره پروتئین وی (WPC) با کیفیت بالا گذاشته است.
مس گینر ویکتور مارتینز، فلسفهای از تغذیه است که توسط خود ویکتور مارتینز طراحی شده است.
مس گینر دنیس جیمز، یک مکمل ورزشی استراتژیک برای افزایش حجم عضلانی است.
کربو رونی کلمن کیسه ای، بر پایه ایده “جذب سریع و فازبندی شده” بنا شده است.
It is actually a nice and helpful piece of info. I am happy that you simply shared this helpful info with us. Please keep us informed like this. Thank you for sharing.
پروتئین وی پریمیوم گلد ویکتور مارتینز، یک مکمل ورزشی پیشرفته و باکیفیت است تحت نظارت مستقیم ویکتور مارتینز.
you are really a good webmaster. The web site loading
velocity is amazing. It sort of feels that you are doing any unique trick.
Also, The contents are masterwork. you have done a wonderful process in this matter!
وی دایماتیز، یک مکمل پروتئینی بسیار محبوب و با کیفیت است.
وی نیتروتک ماسل تک دارای فناوری فیلتراسیون چند فازی است.
کراتین ایس فا نوتریشن، یک پارادایم شیفت (Paradigm Shift) در مهندسی مکملها است.
پروتئین وی ایزوله وایکینگ فورس، نتیجه فرآیندهای پیچیده میکروفیلتراسیون جریان متقاطع (CFM) است.
Статья содержит обновленную информацию и факты, подтвержденные надежными источниками.
I don’t even know how I ended up here, but I thought this post was good. I don’t know who you are but certainly you are going to a famous blogger if you aren’t already 😉 Cheers!
کراتین وایکینگ فورس، با بهرهگیری از تکنولوژیهای پیشرفته فیلتراسیون، محصولی را ارائه میدهد که ذرات آن خیلی ریز هستند.
Автор хорошо подготовился к теме и представил разнообразные факты.
Автор статьи представляет информацию с акцентом на факты и статистику, не высказывая предпочтений.
گینر سریوس مس اپتیموم، یک معماری تغذیهای است که برای شرایط بحرانی کمبود وزن طراحی شده است.
Автор старается оставаться нейтральным, чтобы читатели могли рассмотреть различные аспекты темы.
Я прочитал эту статью с огромным интересом! Автор умело объединил факты, статистику и персональные истории, что делает ее настоящей находкой. Я получил много новых знаний и вдохновения. Браво!
Надеюсь, вам понравятся эти комментарии! Это сообщение отправлено с сайта GoToTop.ee
Hi I am so happy I found your blog page, I really found you by error, while I was looking on Google for something else, Anyways I am here now and would just like to say kudos for a tremendous post and a all round interesting blog (I also love the theme/design), I don’t have time to go through it all at the moment but I have book-marked it and also added your RSS feeds, so when I have time I will be back to read much more, Please do keep up the excellent jo.
Статья представляет объективный анализ проблемы, учитывая разные точки зрения.
Отличная статья! Я бы хотел отметить ясность и логичность, с которыми автор представил информацию. Это помогло мне легко понять сложные концепции. Большое спасибо за столь прекрасную работу!
کراتین مونوهیدرات الیمپ، کراتین نیتروژندار است که به طور طبیعی در کبد و کلیه سنتز میشود.
مس گینر ماسل کور، مفهوم تراکم کالری (Caloric Density) است.
Автор предлагает рациональные и аргументированные выводы на основе представленных фактов.
کراتین مونوهیدرات یو اس ان، با بهرهگیری از تکنولوژیهای پیشرفته فیلتراسیون و میکرونایزیشن، ذرات کراتین را به ابعاد میکرونی خرد کرده است.
Excellent post. Keep posting such kind of information on your page. Im really impressed by it.
Wonderful work! That is the kind of information that should be shared across the web. Disgrace on the seek engines for no longer positioning this post upper! Come on over and talk over with my site . Thank you =)
مس گینر اسکال لبز، یک مکمل غذایی با دانسیته کالری بسیار بالا (High-Calorie Density) است.
Автор статьи поддерживает свои утверждения ссылками на авторитетные источники.
Ahaa, its good discussion on the topic of this post here at this webpage, I have read all that, so now me also commenting at this place.
Мне понравился нейтральный тон статьи, который позволяет читателю самостоятельно сформировать мнение.
کراتین رونی کلمن، یک مکمل ارگوژنیک (افزایشدهنده عملکرد) است که از ۱۰۰٪ کراتین مونوهیدرات خالص تشکیل شده است.
Статья содержит информацию, подкрепленную примерами и исследованиями.
Автор предлагает обоснованные и логические выводы на основе представленных фактов и данных.
Статья представляет все ключевые аспекты темы, обеспечивая при этом достаточную детализацию.
Это помогает читателям получить всестороннее представление о теме без явных предубеждений.
Я хотел бы выразить свою благодарность автору этой статьи за исчерпывающую информацию, которую он предоставил. Я нашел ответы на многие свои вопросы и получил новые знания. Это действительно ценный ресурс!
Статья содержит подробное описание событий и контекста, при этом не выражая пристрастие к какой-либо стороне.
Конечно, вот ещё несколько положительных комментариев на статью. Это сообщение отправлено с сайта https://ru.gototop.ee/
کراتین بادی اتک، یکی از باکیفیتترین و معتبرترین مکملهای کراتین در سطح جهان است.
گینر ایوژن سوپر هیوج، یکی از حرفهایترین و باکیفیتترین مکملهای افزایش وزن و حجم در دنیاست.
Очень хорошо организованная статья! Автор умело структурировал информацию, что помогло мне легко следовать за ней. Я ценю его усилия в создании такого четкого и информативного материала.
Автор старается оставаться нейтральным, что помогает читателям получить полную картину и рассмотреть разные аспекты темы.
پروتئین وی بادی اتک، در هسته مرکزی خود، ترکیبی هوشمندانه از “وی کنسانتره” (Ultra-filtered Whey Concentrate) و “وی هیدرولیزه” (Hydrolyzed Whey) است.
گینر بادی اتک، ترکیبی هوشمندانه از کربوهیدراتهای پیچیده و پروتئینهای چند مرحلهای است.
وی ایزوله بادی اتک، یکی از باکیفیتترین و خالصترین مکملهای پروتئینی در سطح جهان است.
کراتین پلاتینیوم ماسل تک، یکی از خالصترین و پرفروشترین مکملهای کراتین در سطح جهان است.
مس گینر کرتیکال اپلاید نوتریشن، یک فرمولاسیون فوقحرفهای است که فراتر از یک گینر معمولی برای افزایش وزن است.
Автор не высказывает собственных предпочтений, что позволяет читателям самостоятельно сформировать свое мнение.
کراتین بد اس، یک مکمل پیشرفته و قدرتمند است که برخلاف کراتینهای معمولی، ترکیب چندین نوع مختلف کراتین است.
Эта статья – источник ценной информации! Я оцениваю глубину исследования и разнообразие рассматриваемых аспектов. Она действительно расширила мои знания и помогла мне лучше понять тему. Большое спасибо автору за такую качественную работу!
Читатели могут использовать представленную информацию для своего собственного анализа и обдумывания.
Hey there would you mind stating which blog platform you’re using? I’m going to start my own blog in the near future but I’m having a hard time deciding between BlogEngine/Wordpress/B2evolution and Drupal. The reason I ask is because your design and style seems different then most blogs and I’m looking for something completely unique. P.S My apologies for being off-topic but I had to ask!
Мне понравилась логика и четкость аргументации в статье.
I have read so many articles or reviews on the topic of the blogger lovers but this article is really a pleasant piece of writing, keep it up.
Автор предлагает подробное объяснение сложных понятий, связанных с темой.
Amazing! Its really awesome paragraph, I have got much clear idea regarding from this post.
Я хотел бы выразить свою благодарность автору за его глубокие исследования и ясное изложение. Он сумел объединить сложные концепции и представить их в доступной форме. Это действительно ценный ресурс для всех, кто интересуется этой темой.
I am not certain the place you are getting your information, however good topic. I needs to spend a while finding out more or understanding more. Thank you for magnificent info I used to be searching for this info for my mission.
I’m truly enjoying the design and layout of your website. It’s a very easy on the eyes which makes it much more pleasant for me to come here and visit more often. Did you hire out a developer to create your theme? Excellent work!
Это помогает читателям получить полное представление о спорной проблеме.
Информационная статья предлагает взвешенный подход к обсуждаемой теме, аргументируя свои выводы доказательствами и статистикой.
Статья предоставляет информацию, основанную на различных источниках и анализе.
Статья предлагает обширный обзор темы, представляя разные точки зрения и аргументы.
Автор старается сохранить нейтральность и обеспечить читателей информацией для самостоятельного принятия решений.
I real happy to find this internet site on bing, just what I was searching for : D besides saved to favorites.
Статья представляет аккуратный обзор современных исследований и различных точек зрения на данную проблему. Она предоставляет хороший стартовый пункт для тех, кто хочет изучить тему более подробно.
Lovely just what I was looking for.Thanks to the author for taking his time on this one.
Автор старается сохранить нейтральность, чтобы читатели могли сформировать свое собственное понимание представленной информации.
Aw, this was an incredibly nice post. Taking a few minutes and actual effort to make a great article… but what can I say… I procrastinate a lot and never manage to get nearly anything done.
Hello there, simply become aware of your weblog thru Google, and found that it is truly informative. I am gonna be careful for brussels. I will be grateful when you continue this in future. Many other folks might be benefited out of your writing. Cheers!
Автор статьи предоставляет информацию, подкрепленную исследованиями и доказательствами, без выражения личных предпочтений. Это сообщение отправлено с сайта https://ru.gototop.ee/
This is really attention-grabbing, You’re a very skilled blogger. I have joined your rss feed and stay up for in quest of extra of your great post. Also, I’ve shared your web site in my social networks
Amazing! Its really remarkable piece of writing, I have got much clear idea regarding from this piece of writing.
whoah this blog is fantastic i really like studying your articles. Keep up the great work! You recognize, lots of persons are searching around for this info, you can aid them greatly.
Статья содержит достаточно информации для того, чтобы читатель мог сделать собственные выводы.
پروتئین وی امپایر وایکینگ فورس، ریشه در سوئد دارد، کشوری که استانداردهای کنترل کیفیتش در صنایع غذایی و مکمل، زبانزد عام و خاص است.
پروتئین وی بد اس، یک مکمل پروتئینی با کیفیت بالا است که از شیر بهدست میآید
مولتی ویتامین پلاتینیوم ماسل تک، یک مکمل غذایی باکیفیت است که به طور خاص برای ورزشکاران و افرادی که به دنبال بهبود عملکرد ورزشی خود هستند طراحی شده است.
Hello are using WordPress for your site platform? I’m new to the blog world but I’m trying to get started and create my own. Do you require any html coding expertise to make your own blog? Any help would be really appreciated!
Статья содержит достоверные факты и сведения, представленные в нейтральной манере.
Magnificent beat ! I would like to apprentice while you amend your site, how could i subscribe for a blog website? The account aided me a acceptable deal. I had been a little bit acquainted of this your broadcast provided bright clear concept
Hey I know this is off topic but I was wondering if you knew of any widgets I could add to my blog that automatically tweet my newest twitter updates. I’ve been looking for a plug-in like this for quite some time and was hoping maybe you would have some experience with something like this. Please let me know if you run into anything. I truly enjoy reading your blog and I look forward to your new updates.
This is very fascinating, You are a very
skilled blogger. I have joined your feed and stay up for searching for extra
of your wonderful post. Additionally, I have shared your site in my social networks https://instagram.com/
An intriguing discussion is worth comment. I do think that
you ought to publish more on this issue, it may
not be a taboo matter but typically folks don’t discuss these issues.
To the next! Cheers!! https://youtube.com/@Agrafina
کراتین مونوهیدرات گالوانایز، یکی از محبوبترین و مورد مطالعهترین مکملهای ورزشی در جهان است.
Читатели имеют возможность ознакомиться с разными точками зрения и самостоятельно оценить информацию.
مس گینر گالوانایز، یکی از بهترین گزینههای شماست اگر دنبال یک مکمل قوی برای افزایش میزان ماسه و تقویت عضلات هستید.
Статья является исчерпывающим и объективным рассмотрением темы.
Я бы хотел выразить свою благодарность автору этой статьи за его профессионализм и преданность точности. Он предоставил достоверные факты и аргументированные выводы, что делает эту статью надежным источником информации.
پروتئین وی گالوانایز، یک مکمل غذایی محبوب در بین ورزشکاران و افرادی است که به دنبال افزایش مصرف پروتئین خود هستند.
Я нашел эту статью чрезвычайно познавательной и вдохновляющей. Автор обладает уникальной способностью объединять различные идеи и концепции, что делает его работу по-настоящему ценной и полезной.
Because the admin of this website is working, no question very soon it will be well-known, due to its quality contents.
Its like you read my mind! You seem to know a lot about this, like you wrote the book in it or something. I think that you could do with some pics to drive the message home a little bit, but instead of that, this is excellent blog. An excellent read. I’ll certainly be back.
Статья представляет обширный обзор темы и учитывает ее исторический контекст.
Автор статьи умело анализирует сложные концепции и представляет их в понятной форме.
Appreciation to my father who shared with me on the topic of this website, this website is in fact awesome.
I just could not depart your website before suggesting that I extremely loved the standard info a person supply in your visitors? Is gonna be again frequently to check out new posts
Мне понравилась глубина исследования, представленная в статье.
مس گینر روولوشن ماسل اسپرت، یک سیستم کامل برای افزایش کالری دریافتی و پشتیبانی از ریکاوری عضلانی است.
Автор представил четкую и структурированную статью, основанную на фактах и статистике.
Simply want to say your article is as astonishing. The clearness for your put up is just nice and that i could think you are knowledgeable on this subject. Well with your permission let me to grab your RSS feed to keep updated with drawing close post. Thank you a million and please carry on the gratifying work.
Это помогает стимулировать обсуждение и критическое мышление у читателей.
Надеюсь, вам понравятся эти комментарии!
Конечно, вот ещё несколько положительных комментариев на информационную статью: Это сообщение отправлено с сайта GoToTop.ee
Thanks for a marvelous posting! I genuinely enjoyed reading it, you may be a great author. I will make sure to bookmark your blog and may come back in the future. I want to encourage that you continue your great writing, have a nice morning!
Автор статьи представляет данные и факты с акцентом на объективность.
Статья помогла мне лучше понять сложные аспекты темы, с которыми я ранее не сталкивался.
Статья содержит подробное описание событий и контекста, при этом не выражая пристрастие к какой-либо стороне.
وی ایزوله ماسل کور، قلهی تکنولوژی فرآوری پروتئین وی است.
I would like to thank you for the efforts you’ve put in writing this website. I’m hoping to view the same high-grade blog posts from you in the future as well. In fact, your creative writing abilities has encouraged me to get my own website now 😉
Good site you’ve got here.. It’s difficult to find good quality writing like yours these days. I seriously appreciate individuals like you! Take care!!
Мне понравилось, как автор представил информацию в этой статье. Я чувствую, что стал более осведомленным о данной теме благодаря четкому изложению и интересным примерам. Безусловно рекомендую ее для прочтения!
Статья содержит разнообразные факты и аргументы, представленные в объективной манере.
hi!,I like your writing very a lot! percentage we be in contact extra about your post on AOL? I require a specialist on this space to unravel my problem. Maybe that’s you! Looking ahead to peer you.
وی الایت دایماتیز، یک پروتئین ترکیبی هوشمند است.
Автор статьи представляет информацию с акцентом на факты и статистику, не высказывая предпочтений.
Я нашел в статье полезные источники, которые могу изучить для получения дополнительной информации.
Мне понравилось разнообразие и глубина исследований, представленных в статье.
Very good information. Lucky me I discovered your website by chance (stumbleupon). I’ve book marked it for later!
Does your website have a contact page? I’m having a tough time locating it but, I’d like to send you an e-mail. I’ve got some recommendations for your blog you might be interested in hearing. Either way, great website and I look forward to seeing it expand over time.
Howdy would you mind letting me know which web host you’re using? I’ve loaded your blog in 3 different internet browsers and I must say this blog loads a lot quicker then most. Can you recommend a good hosting provider at a honest price? Thanks a lot, I appreciate it!
کراتین بلید اسپورت، با درجه خلوص دارویی تولید میشود.
Статья содержит полезные факты и аргументы, которые помогают разобраться в сложной теме.
I think the admin of this site is truly working hard in support of his website, for the reason that here every information is quality based stuff.
Way cool! Some very valid points! I appreciate you writing this write-up and the rest of the site is extremely good.
Wow that was unusual. I just wrote an incredibly long comment but after I clicked submit my comment didn’t show up. Grrrr… well I’m not writing all that over again. Regardless, just wanted to say superb blog!
Статья предлагает комплексный обзор событий, предоставляя различные точки зрения.
Heya i am for the first time here. I found this board and I find It truly useful & it helped me out much. I hope to give something back and aid others like you aided me.
Увеличение ссылочной массы: Ключевой фактор в росте DR сайта. Увеличение ссылочной массы является неотъемлемой частью стратегии роста DR сайта. Этот процесс требует систематического и длительного подхода, включающего в себя поиск высококачественных ссылок, мониторинг и анализ ссылочной массы, а также адаптацию к постоянно меняющемуся окружению поисковых систем. Понимание этих аспектов и их правильная реализация помогут сайту достичь более высокого DR, что в свою очередь улучшит его ранжирование, привлечет больше органического трафика и повысит успех онлайн-присутствия бизнеса.
Howdy! I’m at work surfing around your blog from my new iphone! Just wanted to say I love reading through your blog and look forward to all your posts! Keep up the outstanding work!
Я хотел бы выразить свою восторженность этой статьей! Она не только информативна, но и вдохновляет меня на дальнейшее изучение темы. Автор сумел передать свою страсть и знания, что делает эту статью поистине уникальной.
Статья предоставляет объективную информацию о теме, подкрепленную различными источниками.
Автор не вмешивается в читателей, а предоставляет им возможность самостоятельно оценить представленную информацию.
obviously like your web-site but you need to take a look at the spelling on quite a few of your posts. Several of them are rife with spelling problems and I to find it very bothersome to inform the reality on the other hand I’ll certainly come back again.
Hey I know this is off topic but I was wondering if you knew of any widgets I could add to my blog that automatically tweet my newest twitter updates. I’ve been looking for a plug-in like this for quite some time and was hoping maybe you would have some experience with something like this. Please let me know if you run into anything. I truly enjoy reading your blog and I look forward to your new updates.
Thanks for sharing your info. I really appreciate your efforts and I am waiting for your next write ups thank you once again.
Highly descriptive blog, I liked that a lot. Will there be a part 2?
پروتئین وی بلید اسپرت، به دلیل سرعت جذب بالا و پروفایل کامل آمینو اسیدی، پادشاه پروتئینها لقب گرفته است.
I have read so many articles on the topic of the blogger lovers but this article is in fact a fastidious post, keep it up.
Мне понравилась глубина исследования, представленная в статье.
Today, I went to the beach front with my kids. I found a sea shell and gave it to my 4 year old daughter and said “You can hear the ocean if you put this to your ear.” She placed the shell to her ear and screamed. There was a hermit crab inside and it pinched her ear. She never wants to go back! LoL I know this is completely off topic but I had to tell someone!
I reckon something truly special in this internet site.
کراتین هیدروکلراید کیجد، یک فرم پیشرفته و مهندسیشده از مکمل کراتین است.