<連載 全3回>
EDCを利用した臨床試験における信頼性調査対応講座 (第1回)
*万が一文中に解釈の間違い等がありましても、当社では責任をとりかねます。
本文書の改訂は予告なく行われることがあります。
CSV規制要件等の改定
1. はじめに
2009年10月19日に開催された「平成21年度GCP研修会」では、演題の一つとして「電子的に収集された臨床試験データに対する信頼性調査の留意点」と題した発表が行われた。
本発表において「EDC調査チェックリスト」の案が紹介され、信頼性調査の内容と事例に基づく留意点の説明があった。
本チェックリストは、既に医薬品医療機器総合機構(以下、PMDA)のホームページで公開されている。
ER/ES指針が、平成17年に発出されてから4年半が経ったが、いよいよ本格的なER/ES指針に基づいた査察が開始されたことになる。
本シリーズでは、3回にわたって、EDCを利用した臨床試験の信頼性調査について、その内容、課題、問題点、対応方法等を考察したい。
今回は、「電子的に収集された臨床試験データに対する信頼性調査の留意点」に関する講演内容の要約を行った。
2.平成21年度GCP研修会
平成21年度GCP研修会は、10月19日に東京、10月23日に大阪で開催された。
そのアジェンダは、図1に示すとおりである。
| 時 間 | 内 容 |
| 13:00~13:10 | 挨拶 (東京)独立行政法人 医薬品医療機器総合機構 理事(技監) 川原 章 (大阪)独立行政法人 医薬品医療機器総合機構 理事長 近藤 達也 |
| 13:10~13:40 | 治験の計画等の届出、治験中の副作用・不具合報告及びIRB登録制度について (東京)PMDA審査マネジメント部 星 順子 (大阪)PMDA審査マネジメント部 北原 淳 |
| 13:40~14:00 | 医療機器GCPの改正及び医療機器の基準適合性調査について (東京)信頼性保証部主任専門員 疋田 理津子 (大阪)信頼性保証部調査専門員 福岡 由紀 |
| 14:00~14:20 | 適合性書面調査の現状と留意点 (東京)信頼性保証部調査専門員 魚津 公一郎 (大阪)信頼性保証部調査専門員 南谷 浄 |
| 14:20~14:40 | GCP実地調査の現状と留意点 (東京)信頼性保証部調査専門員 田中 紀子 (大阪)信頼性保証部調査専門員 田尻 興保 |
| 14:40~15:10 | 治験を行う実施医療機関における留意点 (東京)信頼性保証部調査専門員 城谷 真理 (大阪)信頼性保証部調査専門員 山本 健児 |
| 15:30~16:00 | 電子的に収集された臨床試験データに対する信頼性調査の留意点 (東京)信頼性保証部調査専門員 宇井 英明 (大阪)信頼性保証部調査専門員 福島 弘子 |
| 16:00~16:20 | 医療用後発医薬品に係るGCP実地調査の留意点 (東京)信頼性保証部調査専門員 今泉 克明 (大阪)信頼性保証部調査専門員 川名 純一 |
| 16:20~16:50 | GPMSP/GPSP調査の現状と留意点 (東京)信頼性保証部主任専門員 森山 祐輔 (大阪)信頼性保証部調査専門員 松原 芳幸 |
| 16:50~17:00 | 挨拶 財団法人 日本薬剤師研修センター 専務理事 平山 一男 |
図1 平成21年度GCP研修会アジェンダ
研修会の中で、「電子的に収集された臨床試験データに対する信頼性調査の留意点」という演題があり、EDCを利用した臨床試験における信頼性調査の内容と事例に基づく留意点の説明があった。
本講演の冒頭で「EDC調査に関する検討の位置づけ」として、PMDAの平成21年度計画の紹介があった。(図2)
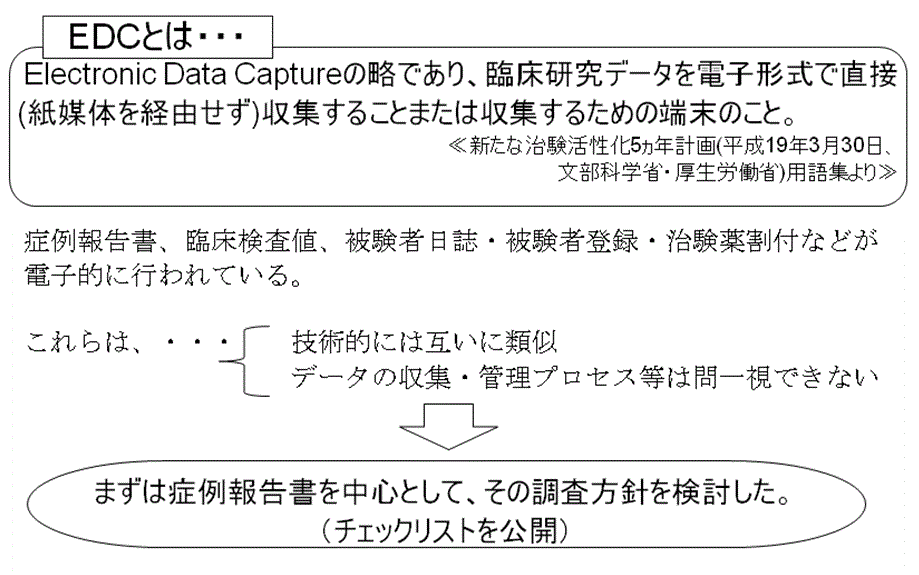 図2 EDC調査に関する検討の位置づけ
図2 EDC調査に関する検討の位置づけ
PMDAの中期計画及び21年度計画において、「急速に進んでいる治験の電子化に対応するため、EDCシステムを中心にシステム調査の検討を進める」とある。
EDCに関する調査において、まず確認すべき事項を定義し、次にシステム調査で確認が可能かどうか、困難をともなうかどうかを検討したとのことであった。
続いて、本講演では、以下の3項目について説明があった。
1) 医療機関で収集される臨床試験データについて
2) 臨床試験データを電子的に取り扱うための関連法令等
3) 電子的に収集されたデータの基準適合性調査と留意点
3. 医療機関で収集される臨床試験データについて
本講演の一番目の内容は、「医療機関で収集される臨床試験データについて」であった。
新たな治験活性化5ヵ年計画の用語集には、EDCとは「Electronic Data Captureの略であり、臨床研究データを電子形式で直接(紙媒体を経由せず)収集することまたは収集するための端末のこと。」とある。(図2 参照)
本発表によると、この用語の定義が意味するところは、かなり広範囲であるという。
例えば、治験依頼者が医療機関から臨床試験データを取得する場合を考えても、症例報告書、中央検査機関による臨床検査値、被験者日誌・被験者登録・治験薬割付など、多くの電子化が行われている。
しかしながら、これらは技術的には互いに類似してはいるものの、データの収集・管理プロセス等は必ずしも同一のものではない。
まずは症例報告書を中心として、その調査方針を検討し、チェックリストを公開したとのことであった。
4. 臨床試験データを電子的に取り扱うための関連法令等
本講演の二番目の内容は、「臨床試験データを電子的に取り扱うための関連法令等」であった。
EDCを利用したとしても、症例報告書の作成については、薬事法施行規則第43条、GCP省令(第47条等)を遵守する必要がある。このことは症例報告書以外のデータでも同様である。
すなわち、基準適合性調査を実施する上での基本的な考え方は変わるものではない。
それらに加えて、電子記録を利用するのであれば、GCP第26条、ER/ES指針およびその上位法令にも留意する必要がある。
4.1 GCP省令第47条 症例報告書等
GCP省令第47条には、症例報告書の作成、署名、修正に関しての規定が記載されている。(図3 参照)
| (症例報告書等) 第47条 治験責任医師等は、治験実施計画書に従って正確に症例報告書を作成し、これに記名なつ印し、又は署名しなければならない。 2 治験責任医師等は、症例報告書の記載を変更し、又は修正するときは、その日付を記載して、これになつ印し、又は署名しなければならない。 3 治験責任医師は、治験分担医師が作成した症例報告書を点検し、内容を確認した上で、これに記名なつ印し、又は署名しなければならない。 |
図3 GCP省令 第47条
治験責任医師等は、症例報告書を作成した際には、記名・捺印または署名を行わなければならない。
また症例報告書の記載を変更したり、修正する際には、その日付を記載したうえで、捺印または署名を行わなければならない。
さらに、治験分担医師が作成した症例報告書であった場合、最終的に治験責任医師がその内容(たとえば、記載事項や修正が適切か等)を確認したうえで記名・捺印または署名を行わなければならない。
また運用通知では、治験責任医師は症例報告書の写しを保存しなければならないとされている。(図4 参照)
| 治験責任医師又は治験分担医師は、症例報告書を治験実施計画書の規定に従って作成し、記名捺印又は署名の上、治験依頼者による治験においては治験依頼者に提出し、自ら治験を実施する者による治験においては自ら治験を実施する者が保存しなければならない。また、治験依頼者に提出した症例報告書の写しを保存するものとする。 |
図4 GCP運用通知 第47条 第1項
4.2 GCP省令第26条 記録の保存等
GCP省令第26条には、記録の保存等に関する規定が記載されている。(図5 参照)
| (記録の保存等) 第26条 治験依頼者は、次に掲げる治験に関する記録(文書及びデータを含む。)を被験薬に係る医薬品についての製造販売の承認を受ける日(第24条第3項の規定により通知したときは、通知した日後3年を経過した日)又は治験の中止若しくは終了の後3年を経過した日のうちいずれか遅い日までの期間適切に保存しなければならない。 1) 治験実施計画書、契約書、総括報告書その他この省令の規定により治験依頼者が作成した文書又はその写し 2) 症例報告書、第32条第6項の規定により通知された文書その他この省令の規定により実施医療機関の長又は治験責任医師等から入手した記録 3) モニタリング、監査その他の治験の依頼及び管理に係る業務の記録(前2号及び第5号に掲げるものを除く。) 4) 治験を行うことにより得られたデータ 5) 第16条第5項に規定する記録 |
図5 GCP省令 第26条
このなかで、治験依頼者は症例報告書を保存しなければならないことが明記されている。
また運用通知において、「記録の保存等 第26条第1項」にかかわる事項として、電子データ処理システムに関する要件があげられている。(図6 参照)
| 3 治験依頼者は、データの処理に当たって、電子データ処理システム(遠隔操作電子データシステムを含む。)を用いる場合には、次の事項を実施しなければならない。電子データ処理システムが、完全性、正確性、信頼性及び意図された性能についての治験依頼者の要件を満たしていることを保証し、文書化すること(すなわちバリデーションされること。)。当該システムを使用するための手順書を整備すること。当該システムが、入力済みのデータを消去することなしに修正が可能で、データ修正の記録をデータ入力者及び修正者が識別されるログとして残せる(すなわち監査証跡、データ入力証跡、修正証跡が残る)ようにデザインされていることを保証すること。データのセキュリティ・システムを保持すること。データのバックアップを適切に行うこと。データの修正を行う権限を与えられた者の名簿を作成し、管理すること。盲検化が行われている場合には、盲検性が保持されるようにすること。 |
図6 GCP運用通知 記録の保存等 第26条第1項
これらは、ICH-GCPの邦訳であり、平成9年3月27日に中薬審答申第40号として発表されたものと同一である。
このなかで「遠隔操作電子データシステム」とは、EDCシステムを指すと考えられる。
1) バリデーション
バリデーションは、コンピュータシステムを使用する際の必須要件である。
2) 手順書
当該システムを使用するための手順書を整備しなければならない。手順書は、システムの操作手順書のみではなく、セキュリティ管理に関する手順書、監査証跡確認手順書、電磁的記録媒体の保存性に関する手順書、バックアップ/リカバリ手順書等の、電子記録・電子署名を使用する上で必要となる手順書を含む。
3) 監査証跡
当該システムは、監査証跡が自動的に記録されるように設計されていなければならない。
監査証跡は、改ざんを発見するために必須の機能である。
4) セキュリティ
データのセキュリティ・システムを保持しなければならない。
セキュリティは、なりすましや改ざんを防止するために必要である。
セキュリティには、物理的セキュリティ、論理的セキュリティ、ネットワークセキュリティ、人的セキュリティなどが存在する。
物理的セキュリティは、サーバルームには施錠を行い、無用な者を入室させないというものがあげられる。またEDCシステムの端末を、往来の多い場所を避け、研究室などの施錠できる場所に設置するなども考えられる。
論理的セキュリティとは、パスワードを利用し、アクセス制限や権限設定を行うことを指す。また一定時間入力がなかった場合に、自動的にスクリーンセーバーなどを起動させることも有効である。
ネットワークセキュリティは、ファイヤーウォールやウィルス駆除ソフトウェアなどの導入を指す。またウィニー(Winny)などの不適切なソフトウェアがインストールされている場合、情報漏えいにつながる危険性がある。
人的セキュリティは、パスワードを他人に教えない、紙などに記載しておかないなどである。これらを周知徹底するために、ユーザ教育は重要である。
5) バックアップ
既に解説した通り、バックアップは規制要件であり、真正性の要件である。
データのバックアップを適切に行うことも必要であるが、災害時等に適切に回復(リカバリ)できることも重要である。
6) データの修正を行う権限を与えられた者の名簿
データの修正を行う権限を与えられた者は、電子記録の改ざんが可能になる。したがって、適宜、誰が修正権限を持っているのかを適切に管理しなければならない。
年度初めなどには、治験責任医師等の異動が多くなる。当該治験責任医師等が、当該治験を外れた場合、ユーザ権限をすみやかに無効化しなければならない。
また最終の被験者が治験を終えた際(LPO:Last Patient Out)の際などにも、必要のなくなった権限は無効化しておかなければならない。
これらアクセス権限等を、アカウント管理表等を作成し、常時管理しなければならない。
7) 盲検性の保持
EDCシステムを利用すると、症例データの集積にともない、リアルタイムに治験中のデータの抽出や中間解析が可能となる。
盲検化された試験の場合、中間解析等を行うことは、盲検性の保持の面から極めて不適切である。
5. 電子的に収集されたデータの基準適合性調査と留意点
本講演の三番目の内容は、「電子的に収集されたデータの基準適合性調査と留意点」であり、多くの聴衆が関心を持つ内容である。
講演内容によると、平成20年度以降、10社以上(20申請品目以上)でEDCを利用した試験を含む申請品目の適合性調査がPMDAによって実施されてきたという。ちなみに平成19年度までは2申請品目だったとのことであった。
PMDAでは、平成 21年3月に、EDCデータに対する調査すべき事項を「EDC調査チェックリスト(案)」としてまとめた。
その後、平成21年5月から8月にかけて、「EDC調査チェックリスト(案)」を用いてパイロット調査を行ってきたという。
このチェックリストは、既にPMDAのHPで公開されている。
http://www.pmda.go.jp/operations/shonin/outline/shinrai/checklist.html
本チェックリストは、Wordが治験依頼者用、pdfが医療機関用となっている。
ちなみに、このようなチェックリストを課長通知等ではなく、PMDAのホームページで公開するという意義は、常に見直しと改定が行われることを意味すると筆者は理解している。
本チェックリストの内容は、図7に示す通りとなっている。
| (平成21年10月現在)EDC調査チェックリスト (治験依頼者用)1. システムの概要について 2. EDCシステム運用に関する治験依頼者の組織・体制・委託状況等 3. バリデーションについて 4. ユーザー管理について 5. データの保存 6. 電子症例報告書の作成、修正及び署名についてEDC調査チェックリスト(医療機関用)1. ユーザー管理について 2. データの保存 |
図7 EDC調査チェックリストの構成
本演題では、チェックリストの内容の紹介とともに、調査の内容と事例に基づく留意点の説明があった。
チェックリストは、本来の調査項目に加えて、EDCを使用した場合に追加となる差分の項目のみが記載されていることの説明があった。
監査証跡のバリデーションに関しての調査項目が存在することが特徴的である。
また治験依頼者の要件を満たしていることを保証する文書の調査も加わった。
さらに各種手順書に関して、当該治験に該当するものの作成日を調査することになっている。
バリデーションに関しては、ASPを使用するケースを例とし、開発者、ASP、治験依頼者の関係を図で示し、このパターンが最も複雑になること、しかしながら最終責任は治験依頼者にあることの説明があった。
バリデーションについての深い調査は行っていないが、要検討項目であることが付け加えられた。
5.1 治験依頼者に関する留意点
治験依頼者に関する留意点として、以下のような事例等が報告された。
1) ユーザ管理において、システムが万全であっても、使う側が適切に運用しなければならないこと。
2) 教育訓練を行っていても、ID、Passwordの発行時に、受け手(医療機関)が理解できていない事例が発生している。
3) データの保存に関する留意事項として、多くはpdfで作成されているが、治験責任医師の署名後に修正が発生した場合、最終版が保存されていない事例があった。
これは第26条の適切な保存の要件に抵触するものである。
4) 電子症例報告書の作成、修正および署名について、本来(紙ベースのCRFの場合)は医療機関が責任を持つべきであるが、EDCを利用する場合は、治験依頼者がそれらの環境を準備するため、治験依頼者側での調査を行っている。
医療機関においては、紙ベースの調査と変わらないため、本項目の医療機関用チェックリストは作成していない。
5) 電子症例報告書は、監査証跡を含めて完全といえる。
6) pdf化する際に特殊文字などによる文字化け、表示のズレ等が発生している事例がある。
7) EDCでは、権限設定により、エラーを防ぐことができることがある。
例えば、最終確認署名は治験責任医師のみが可能となるようにし、治験分担医師はダメとするなど。
あくまでも責任は医療機関側にあるが、色んなシステムがあり、覚えきれないので、(機能を工夫するなど)できることをやってもらいたい。
5.2 医療機関に関する留意点
医療機関に関する留意点として、以下のような事例等が報告された。
1) ID・パスワードが、医療機関毎にひとつ(共有)と勘違いしたケースがある。
2) 資格のない者に入力を代行させている事例がある。これはEDCを利用しているかどうか以前の問題である。
3) 利用権限が同等以上であるユーザであれば、代わりに入力してもかまわないといった勘違い事例があった。
4) 医療機関が電子症例報告書(写)を受け取る際、治験依頼者が作成したものであるから大丈夫であろうとする考え方は良くない。
悪意はなくとも人為的ミスはあり得る。電子症例報告書(写)を受領する際に、内容を確認すること。
記録の保存責任はあくまでも医療機関にある。受領書にその旨記載されていると思う。受領書にサインしたことによって内容に責任を持つことになる。
6. おわりに
今回は、平成21年度 GCP研修会における、「電子的に収集された臨床試験データに対する信頼性調査の留意点」に関する講演内容の要約を試みた。
本講演では、事例に基づいた留意点が紹介されたが、これはあくまでもPMDAの査察官が見付け出せた内容に基づくものである。
今後も調査が継続され、本チェックリストは事例に基づき改定されるものであろうと推察する。
しかしである。本来、規制当局は警察官のような立場であっては困る。
たまたま調査で見付け出すことができた事例に関しては、指摘を行い改善が図られるが、そうでない事項においては瑕疵が残存してしまうからである。
例えばスピード違反の場合、警察官が目撃している場合には取り締まられるが、そうでない場合には違反が見逃されることになる。指摘した場合のみ是正が行われるといった方法では、根本的な品質保証は図れない。
かつてのFDAもそうであった。1997年に成立した、FDA近代化法以降、FDAは査察方針について見直しと改善を行った。
FDAが発行する最近のワーニングレターには、次のような一節が加えられることが多い。
「このワーニングレターは、貴社におけるすべての違反を列記したものではないことを、承知して頂きたい。該当する法律やFDAの規制要件を遵守することの保証に関する責任は貴社にある。」
問題は、日本では当局がEDCに関する規制要件を発表していないことである。規制要件がなければ、遵守すべき事項が不明確である。したがって、適合性を持ったSOPの作成もできないことになる。
本来チェックリストは、拠り所とする規制要件に基づいて作成されるべきである。
GCP省令26号や、ER/ES指針では不十分である。
またCSVに関する規制要件も発表されていない中で、バリデーションを実施しているかといった調査も無理があるように思える。
このような方法では、調査がうわべだけのものになってしまう。
FDAが、2007年5月に発行した「Guidance for Industry Computerized Systems Used in Clinical Investigations」では、背景として次のように述べている。
「FDAが臨床試験データを受領するかどうかは、オンサイトの査察や監査中に、データの品質や完全性を確認できるかどうかというFDAの能力にかかっている。」
奇しくも、平成19年12月21日に日本製薬工業協会 医薬品評価委員会が開催した「臨床試験データの電子的取得に関するガイダンス」説明会において、PMDA 新薬審査第2部(当時)の井本昌克氏が講演し、「EDCを利用した試験成績が受け入れ可能か不明」と述べている。
次回は、「EDC調査チェックリスト(案)」の内容を筆者なりに解釈し、製薬企業における対応の方法等について考察を行ってみたい。
参考
1) 「平成21年度 GCP研修会 講演要旨集」 平成21年10月19日 財団法人 日本薬剤師研修センター
2) 「医薬品等の承認又は許可等に係る申請等における電磁的記録及び電子署名の利用について」平成17年4月1日 薬食発第0401022号
3) 「臨床試験データの電子的取得に関するガイダンス」平成19年11月1日 日本製薬工業協会 医薬品評価委員会

EDCを利用した治験講座 その1
EDCを利用した治験講座 その2
EDCを利用した治験講座 その3
For most recent news you have to go to see the web and on web
I found this web page as a most excellent website
for latest updates.
Simply want to say your article is as amazing. The clarity in your post is just great and i could assume you are an expert on this subject. Well with your permission allow me to grab your feed to keep updated with forthcoming post. Thanks a million and please carry on the enjoyable work.
I am glad to be a visitant of this gross site! , regards for this rare info ! .
I genuinely enjoy studying on this site, it contains fantastic blog posts.
Pretty great post. I just stumbled upon your blog and wished to mention that I’ve truly loved browsing your weblog posts. In any case I will be subscribing in your feed and I am hoping you write again very soon!
Tonic Greens: An Overview Introducing Tonic Greens, an innovative immune support supplement meticulously crafted with potent antioxidants, essential minerals, and vital vitamins.
I’ve recently started a website, the information you offer on this website has helped me tremendously. Thanks for all of your time & work. “Everyone is responsible and no one is to blame.” by Will Schutz.
Great write-up, I’m regular visitor of one’s web site, maintain up the excellent operate, and It is going to be a regular visitor for a lengthy time.
Hi there this is kinda of off topic but I was wondering if blogs use WYSIWYG editors or if you have to manually code with HTML. I’m starting a blog soon but have no coding experience so I wanted to get advice from someone with experience. Any help would be enormously appreciated!
I used to be able to find good advice from your blog posts.
You are my inspiration , I have few blogs and rarely run out from to brand : (.
I see something really special in this site.
wonderful post, very informative. I wonder why the other specialists of this sector don’t notice this. You must continue your writing. I’m sure, you have a great readers’ base already!
Rattling informative and wonderful anatomical structure of subject material, now that’s user friendly (:.
I simply couldn’t depart your website prior to suggesting that I actually enjoyed the usual information a person supply in your visitors? Is going to be back regularly in order to check out new posts
I was examining some of your articles on this site and I believe this site is really instructive! Continue posting.
Somebody essentially help to make seriously articles I would state. This is the very first time I frequented your web page and thus far? I amazed with the research you made to make this particular publish extraordinary. Wonderful job!
The next time I read a blog, I hope that it doesnt disappoint me as much as this one. I mean, I know it was my choice to read, but I actually thought youd have something interesting to say. All I hear is a bunch of whining about something that you could fix if you werent too busy looking for attention.
Yay google is my queen helped me to find this outstanding web site! .
Hello! Do you know if they make any plugins to assist with SEO?
I’m trying to get my blog to rank for some targeted keywords but I’m not
seeing very good success. If you know of any please
share. Appreciate it!
I’d like to thank you for the efforts you’ve put
in penning this site. I really hope to check out the same high-grade content
by you in the future as well. In fact, your creative writing abilities has inspired me to get my very own website now 😉
You should be a part of a contest for one of the best websites on the web.
I will recommend this website!
Saved as a favorite, I love your web site!
I love your blog.. very nice colors & theme. Did you make
this website yourself or did you hire someone to do it for you?
Plz answer back as I’m looking to design my own blog and would
like to know where u got this from. cheers
Hi there, I enjoy reading through your post. I like to write a little comment
to support you.
Hi! Quick question that’s completely off topic. Do you know how to make your site mobile friendly?
My website looks weird when viewing from my iphone. I’m
trying to find a template or plugin that might be able to correct this problem.
If you have any recommendations, please share.
Appreciate it!
Hello to every , because I am genuinely eager of reading this blog’s post to be updated daily.
It contains good data.
Thanks for one’s marvelous posting! I genuinely enjoyed reading it,
you could be a great author.I will ensure that
I bookmark your blog and will come back someday. I want to
encourage you to ultimately continue your great
work, have a nice evening!
I have been exploring for a little for any high-quality articles or weblog posts in this kind of
area . Exploring in Yahoo I at last stumbled upon this web site.
Reading this information So i am satisfied to express that I’ve a very excellent uncanny feeling
I came upon exactly what I needed. I so much indisputably will make sure to do not put out of your mind this site and
provides it a look regularly.
Somebody essentially assist to make severely articles I might
state. This is the very first time I frequented your website page and thus far?
I surprised with the research you made to make this actual put up extraordinary.
Excellent process!
Have you ever thought about publishing an ebook or guest authoring on other websites?
I have a blog based upon on the same information you discuss
and would love to have you share some stories/information. I know my visitors would appreciate your work.
If you are even remotely interested, feel free to shoot me an e-mail.
Unquestionably believe that which you stated. Your favourite reason appeared to be on the internet the simplest thing to
consider of. I say to you, I certainly get irked while other folks think about issues that they just don’t
realize about. You managed to hit the nail upon the highest as neatly as defined out the entire thing
without having side-effects , other people could
take a signal. Will probably be back to get more.
Thanks
whoah this blog is wonderful i like studying your posts. Stay
up the great work! You already know, many individuals are looking round for this info,
you can aid them greatly.
What’s Going down i’m new to this, I stumbled upon this I’ve found It absolutely useful
and it has helped me out loads. I’m hoping to contribute & aid different users like its helped me.
Great job.
Every weekend i used to visit this web page, because i want enjoyment, since this this web page conations truly pleasant funny material too.
It’s amazing designed for me to have a web site, which is valuable for my
know-how. thanks admin
I’m gone to say to my little brother, that he should also pay
a visit this weblog on regular basis to get updated from
hottest news.
Hello, I desire to subscribe for this blog to take latest updates, so where can i do it please assist.
Exceptional post however , I was wanting to know
if you could write a litte more on this subject? I’d be very grateful if you could elaborate a
little bit more. Appreciate it!
I know this if off topic but I’m looking into starting my own blog and was curious what all is required to get setup?
I’m assuming having a blog like yours would cost a pretty penny?
I’m not very web savvy so I’m not 100% certain. Any tips or advice
would be greatly appreciated. Thanks
At this time I am going to do my breakfast, after having my breakfast
coming yet again to read other news.
For the reason that the admin of this website is working,
no hesitation very rapidly it will be renowned, due to its feature contents.
I really like your blog.. very nice colors & theme. Did
you make this website yourself or did you hire someone to do it for you?
Plz respond as I’m looking to create my own blog and would like to find out where u got this
from. kudos
Hi there to all, how is everything, I think every one is
getting more from this web page, and your views
are pleasant in favor of new viewers.
you’re actually a excellent webmaster. The web site loading speed is
incredible. It seems that you are doing any unique trick.
In addition, The contents are masterwork. you have done a magnificent process on this matter!
Hi there, just became aware of your blog through Google, and found that it is truly informative.
I am going to watch out for brussels. I will appreciate
if you continue this in future. Numerous people will be benefited from your writing.
Cheers!
I have been browsing on-line greater than three hours nowadays, but I by no means found any interesting
article like yours. It’s lovely price sufficient for me.
In my view, if all web owners and bloggers made good
content material as you did, the web will be much more helpful than ever before.
Thanks for sharing such a good thought, paragraph is good, thats why i have read it entirely
Great blog you have here but I was wondering if you knew of any user discussion forums that cover
the same topics discussed in this article? I’d really like
to be a part of group where I can get suggestions from other experienced people that share the same interest.
If you have any suggestions, please let me know.
Appreciate it!
Hey There. I found your weblog the use of msn. This is a really smartly
written article. I’ll be sure to bookmark it and come back to read more of your useful info.
Thank you for the post. I’ll certainly return.
Howdy! This blog post couldn’t be written much better!
Going through this article reminds me of my previous roommate!
He constantly kept talking about this. I’ll send this post to him.
Pretty sure he will have a great read. Thanks for sharing!
This design is wicked! You obviously know how to keep a reader amused.
Between your wit and your videos, I was almost moved to start
my own blog (well, almost…HaHa!) Fantastic job. I really enjoyed what you had
to say, and more than that, how you presented it. Too cool!
continuously i used to read smaller articles or reviews that also clear their motive, and that is also happening with this
piece of writing which I am reading at this time.
Good day! This is my first comment here so I just wanted to give
a quick shout out and say I really enjoy reading your
articles. Can you recommend any other blogs/websites/forums that deal with the same topics?
Thanks!
Nice post. I learn something new and challenging on sites I stumbleupon every day.
It’s always helpful to read content from other writers
and practice a little something from their web sites.
It’s truly very difficult in this full of activity life to listen news on Television, thus I simply use world wide web for that purpose, and obtain the latest news.
My relatives always say that I am killing my time here at net, but I know
I am getting experience daily by reading thes pleasant articles
or reviews.
It’s an awesome piece of writing for all the online people;
they will take advantage from it I am sure.
An intriguing discussion is worth comment.
There’s no doubt that that you need to write more about this subject
matter, it may not be a taboo matter but generally people do not talk about these subjects.
To the next! Best wishes!!
I think this is one of the most vital info for me. And i’m
glad reading your article. But want to remark on few general things, The site style is wonderful, the articles
is really nice : D. Good job, cheers
This design is steller! You certainly know how to keep a reader entertained.
Between your wit and your videos, I was almost moved to start my own blog (well,
almost…HaHa!) Great job. I really enjoyed what you had to say, and more than that, how you presented it.
Too cool!
Fine way of describing, and nice piece of writing to obtain information on the topic of my presentation topic,
which i am going to deliver in institution of higher education.
This page certainly has all the info I wanted concerning
this subject and didn’t know who to ask.
What’s up, everything is going perfectly here and ofcourse every one is sharing
facts, that’s really excellent, keep up writing.
I have been exploring for a little bit for any high-quality articles or weblog posts in this sort of area . Exploring in Yahoo I finally stumbled upon this site. Reading this info So i am glad to express that I have a very good uncanny feeling I found out just what I needed. I so much definitely will make sure to don’t forget this website and give it a look on a continuing basis.
Does your site have a contact page? I’m having problems locating it but,
I’d like to send you an e-mail. I’ve got some ideas for your blog you might be interested in hearing.
Either way, great site and I look forward to seeing it improve over time.
May I simply say what a comfort to find someone who genuinely understands what they’re discussing on the
web. You definitely understand how to bring an issue to light and make it important.
More people have to check this out and understand this side of the story.
I was surprised you’re not more popular because you most certainly have the gift.
Hi there mates, how is everything, and what you would like to
say on the topic of this piece of writing, in my view
its truly amazing in support of me.
bookmarked!!, I love your site!
Great website. Lots of useful info here. I’m sending it to a few friends ans also sharing in delicious. And certainly, thanks for your effort!
Way cool! Some extremely valid points! I appreciate you penning this article
and the rest of the website is extremely good.
I know this site provides quality depending posts and extra material,
is there any other website which presents these kinds of stuff in quality?
It’s the best time to make a few plans for the long run and it
is time to be happy. I have learn this post and if I may just I want to recommend you few attention-grabbing issues or suggestions.
Perhaps you could write subsequent articles referring to this article.
I wish to learn more things about it!
This article is truly a nice one it assists new the
web users, who are wishing for blogging.
It’s awesome how magnificent this site is Get more info
Absolutely pent subject material, appreciate it for information. “The bravest thing you can do when you are not brave is to profess courage and act accordingly.” by Corra Harris.
Some genuinely interesting details you have written.Assisted me a lot, just what I was looking for : D.
I think this internet site has some really superb information for everyone : D.
That’s a nice site that we could appreciate Get more info
That’s a nice site that we could appreciate Get more info
That’s a nice site that we could appreciate Get more info
That’s a nice site that we could appreciate Get more info
That’s a nice site that we could appreciate Get more info
That’s a nice site that we could appreciate Get more info
That’s a nice site that we could appreciate Get more info
That’s a nice site that we could appreciate Get more info
That’s a nice site that we could appreciate Get more info
That’s a nice site that we could appreciate Get more info
I blog often and I seriously thank you for your content.
Your article has truly peaked my interest. I will take a note of your site
and keep checking for new information about once per week.
I subscribed to your RSS feed too.
Thanks for the marvelous posting! I seriously enjoyed reading it, you are a great
author. I will be sure to bookmark your blog and definitely will come back at some point.
I want to encourage you to ultimately continue your great posts,
have a nice day!
Howdy! Do you know if they make any plugins
to help with Search Engine Optimization? I’m trying to get my blog to rank for some
targeted keywords but I’m not seeing very good success.
If you know of any please share. Many thanks!
An outstanding share! I’ve just forwarded this onto a friend who had
been conducting a little research on this. And he in fact bought me breakfast because I discovered it for him…
lol. So let me reword this…. Thanks for the meal!!
But yeah, thanx for spending the time to talk about this subject here on your site.
First of all I would like to say terrific blog! I had a quick question which I’d like to ask if you don’t mind.
I was curious to find out how you center yourself and
clear your thoughts before writing. I’ve had a tough time clearing my
mind in getting my thoughts out there. I do take pleasure in writing but it just seems like the first
10 to 15 minutes tend to be wasted simply just trying to figure out how to begin.
Any ideas or tips? Kudos!
Howdy! I could have sworn I’ve been to this site before but after reading through some of the post I realized it’s new to me.
Anyways, I’m definitely glad I found it and I’ll be bookmarking and checking back
frequently!
I am not sure where you are getting your information, but good topic.
I needs to spend some time learning much more or understanding more.
Thanks for wonderful information I was looking for this information for my mission.
Hola! I’ve been following your blog for some time now and
finally got the courage to go ahead and give you a shout out from
New Caney Texas! Just wanted to mention keep up the fantastic work!
It’s an amazing piece of writing designed for all the internet people; they will take advantage from it I
am sure.
Simply want to say your article is as amazing.
The clarity in your post is simply great and i can assume you
are an expert on this subject. Fine with your permission let me to grab your RSS feed to
keep up to date with forthcoming post. Thanks a
million and please keep up the gratifying work.
Exceptional post but I was wondering if you could write
a litte more on this topic? I’d be very grateful if
you could elaborate a little bit more. Thank you!
I don’t even understand how I stopped up right here, but
I thought this publish was great. I do not realize who you might be however definitely you’re going to
a famous blogger for those who aren’t already. Cheers!
buy viagra online
Влияние увеличенной посещаемости на монетизацию сайта. Для владельцев сайтов, которые зарабатывают на рекламе или партнёрских программах, увеличение посещаемости может привести к повышению доходов. Сервис SiteGoToTop.com может стать эффективным инструментом для увеличения числа кликов по рекламным объявлениям или партнёрским ссылкам. Таким образом, рост трафика способствует увеличению прибыли.
Your style is unique compared to other folks I have read stuff from.
Thanks for posting when you’ve got the opportunity,
Guess I’ll just bookmark this page.
Pretty nice post. I just stumbled upon your blog and wanted to say that I’ve truly enjoyed browsing your blog posts. In any case I’ll be subscribing to your rss feed and I hope you write again very soon!
Я оцениваю объективность и сбалансированность аргументации в статье.
Статья является информативной и предоставляет различные факты и аргументы.
Thanks for the good writeup. It in reality used to be a entertainment account it. Look complicated to far brought agreeable from you! However, how can we keep in touch?
Хорошая статья, в которой представлены факты и документированные данные.
Spot on with this write-up, I actually feel this website needs a great deal more attention. I’ll probably be back again to read more, thanks for the info!
Статья предлагает разнообразные подходы к решению проблемы и позволяет читателю выбрать наиболее подходящий для него.
Hi! I just wanted to ask if you ever have any issues with hackers? My last blog (wordpress) was hacked and I ended up losing months of hard work due to no backup. Do you have any solutions to stop hackers?
Admiring the time and effort you put into your site and in depth information you provide. It’s great to come across a blog every once in a while that isn’t the same old rehashed material. Excellent read! I’ve bookmarked your site and I’m including your RSS feeds to my Google account.
Я нашел эту статью чрезвычайно познавательной и вдохновляющей. Автор обладает уникальной способностью объединять различные идеи и концепции, что делает его работу по-настоящему ценной и полезной.
Я впечатлен этой статьей! Она не только информативна, но и вдохновляющая. Мне понравился подход автора к обсуждению темы, и я узнал много нового. Огромное спасибо за такую интересную и полезную статью!
Влияние увеличенной посещаемости на монетизацию сайта. Для владельцев сайтов, которые зарабатывают на рекламе или партнёрских программах, увеличение посещаемости может привести к повышению доходов. Сервис SiteGoToTop.com может стать эффективным инструментом для увеличения числа кликов по рекламным объявлениям или партнёрским ссылкам. Таким образом, рост трафика способствует увеличению прибыли.
Я оцениваю четкую структуру статьи, которая помогает организовать мысли и понять ее содержание.
Автор не высказывает собственных предпочтений, что позволяет читателям самостоятельно сформировать свое мнение.
Автор представляет информацию в увлекательном и легко усваиваемом формате.
Это позволяет читателям получить разностороннюю информацию и самостоятельно сделать выводы.
It’s very simple to find out any topic on net as compared to books, as I found this article at this web page.
Автор предоставляет разнообразные источники для более глубокого изучения темы.
My family all the time say that I am wasting my time here at net, but I know I am getting familiarity every day by reading such nice articles.
Your style is really unique compared to other people I have read stuff from. Many thanks for posting when you’ve got the opportunity, Guess I will just book mark this web site.
excellent points altogether, you simply gained a logo new reader. What would you recommend about your publish that you simply made a few days in the past? Any sure?
I used to be able to find good info from your content.
Эта статья оказалась исключительно информативной и понятной. Автор представил сложные концепции и теории в простой и доступной форме. Я нашел ее очень полезной и вдохновляющей!
Я хотел бы выразить свою благодарность автору этой статьи за исчерпывающую информацию, которую он предоставил. Я нашел ответы на многие свои вопросы и получил новые знания. Это действительно ценный ресурс!
Статья содержит актуальную информацию по данной теме.
Does your site have a contact page? I’m having a tough time locating it but, I’d like to shoot you an e-mail. I’ve got some ideas for your blog you might be interested in hearing. Either way, great blog and I look forward to seeing it improve over time.
Статья содержит систематическую аналитику темы, учитывая разные аспекты проблемы.
Статья представляет различные аспекты темы и помогает получить полную картину.
Статья содержит актуальную информацию, которая помогает понять сложность и важность проблемы.
Hi! Do you use Twitter? I’d like to follow you if that would be okay. I’m undoubtedly enjoying your blog and look forward to new updates.
Я оцениваю объективность автора и его стремление представить разные точки зрения на проблему.
Good replies in return of this issue with solid arguments and describing the whole thing on the topic of that.
Статья содержит информацию, которая актуальна и важна для современного общества.
This is my first time go to see at here and i am really pleassant to read all at one place.
Я нашел в статье несколько полезных советов.
Статья содержит разнообразные точки зрения, представленные в равной мере.
Я не могу не отметить качество исследования, представленного в этой статье. Автор использовал надежные источники и предоставил нам актуальную информацию. Большое спасибо за такой надежный и информативный материал!
Статья представляет анализ разных точек зрения на проблему, что помогает читателю получить полное представление о ней.
An intriguing discussion is worth comment. I do believe that you should publish more on this subject, it might not be a taboo matter but usually people do not discuss these issues. To the next! Cheers!!
Great article! We are linking to this great content on our website. Keep up the good writing.
Автор предлагает практические советы, которые читатели могут использовать в своей повседневной жизни.
Normally I do not learn article on blogs, however I wish to say that this write-up very pressured me to check out and do so! Your writing taste has been amazed me. Thank you, very nice post.
Статья содержит информацию, основанную на достоверных источниках и экспертных мнениях.
I do not even know how I ended up here, but I thought this post was great. I do not know who you are but definitely you are going to a famous blogger if you are not already 😉 Cheers!
Радует объективность статьи, автор старается представить информацию без сильной эмоциональной окраски.
Thanks a bunch for sharing this with all of us you really realize what you are speaking approximately! Bookmarked. Please also discuss with my website =). We will have a hyperlink change agreement among us
Надеюсь, что эти дополнительные комментарии принесут ещё больше позитивных отзывов на информационную статью!
Хорошая статья, которая основана на фактах и не сторонится от нейтральности.
Статья представляет обобщенный взгляд на проблему, учитывая ее многогранные аспекты.
This is a really good tip particularly to those new to the blogosphere. Short but very accurate info… Many thanks for sharing this one. A must read article!
Очень интересная исследовательская работа! Статья содержит актуальные факты, аргументированные доказательствами. Это отличный источник информации для всех, кто хочет поглубже изучить данную тему.
Статья содержит релевантную информацию, актуальную для современной обстановки.
Статья содержит анализ преимуществ и недостатков разных решений проблемы, помогая читателю принять информированное решение.
Asking questions are in fact fastidious thing if you are not understanding anything totally, except this paragraph gives nice understanding yet.
Автор статьи умело анализирует сложные концепции и представляет их в понятной форме.
Автор статьи предоставляет информацию, основанную на различных источниках и экспертных мнениях.
Автор статьи представляет информацию с акцентом на факты и статистику, не высказывая предпочтений.
Автор статьи предоставляет информацию, основанную на различных источниках и экспертных мнениях.
Очень понятная и информативная статья! Автор сумел объяснить сложные понятия простым и доступным языком, что помогло мне лучше усвоить материал. Огромное спасибо за такое ясное изложение! Это сообщение отправлено с сайта https://ru.gototop.ee/
This post will assist the internet users for setting up new webpage or even a blog from start to end.
Это помогает читателям получить объективное представление о рассматриваемой теме.
I am extremely impressed with your writing skills as well as with the layout on your blog. Is this a paid theme or did you customize it yourself? Either way keep up the nice quality writing, it’s rare to see a great blog like this one these days.
Just want to say your article is as astounding. The clearness in your put up is just great and i can assume you are an expert on this subject. Well along with your permission let me to clutch your RSS feed to keep up to date with impending post. Thanks a million and please carry on the rewarding work.
Я нашел эту статью чрезвычайно познавательной и вдохновляющей. Автор обладает уникальной способностью объединять различные идеи и концепции, что делает его работу по-настоящему ценной и полезной.
Эта статья является примером качественного исследования и профессионализма. Автор предоставил нам широкий обзор темы и представил информацию с точки зрения эксперта. Очень важный вклад в популяризацию знаний!
Информационная статья содержит полезный контент и представляет различные аспекты обсуждаемой темы.
Статья предоставляет информацию из разных источников, обеспечивая балансированное представление фактов и аргументов.
Я прочитал эту статью с огромным интересом! Автор умело объединил факты, статистику и персональные истории, что делает ее настоящей находкой. Я получил много новых знаний и вдохновения. Браво!
Это помогает читателям получить полное представление о сложности и многогранности обсуждаемой темы.
Я оцениваю объективность и сбалансированность аргументации в статье.
Статья представляет различные аспекты темы и помогает получить полную картину.
You’re so awesome! I do not suppose I have read a single thing like this before. So nice to find another person with some unique thoughts on this issue. Really.. thanks for starting this up. This site is one thing that is needed on the internet, someone with a bit of originality!
Автор старается подойти к теме без предубеждений.
Это позволяет читателям самостоятельно оценить и проанализировать информацию.
I really like your blog.. very nice colors & theme. Did you create this website yourself or did you hire someone to do it for you? Plz respond as I’m looking to design my own blog and would like to find out where u got this from. thanks
Я оцениваю четкую структуру статьи, которая делает ее легко читаемой и понятной.
It’s an remarkable article for all the internet visitors; they will obtain benefit from it I am sure.
Have you ever considered about including a little bit more than just your articles? I mean, what you say is valuable and everything. However think of if you added some great pictures or videos to give your posts more, “pop”! Your content is excellent but with images and clips, this site could certainly be one of the best in its field. Superb blog!
Я хотел бы поблагодарить автора этой статьи за его основательное исследование и глубокий анализ. Он представил информацию с обширной перспективой и помог мне увидеть рассматриваемую тему с новой стороны. Очень впечатляюще!
Надеюсь, вам понравятся и эти комментарии!
Автор представляет сложные понятия в понятной и доступной форме, что erleichtert das Verständnis.
I know this website gives quality dependent articles or reviews and additional material, is there any other website which offers these kinds of data in quality?
Hi mates, how is all, and what you desire to say about this post, in my view its in fact awesome in favor of me.
Автор представляет сложные концепции и понятия в понятной и доступной форме.
Everything posted was actually very reasonable. But, what about this? suppose you added a little content? I mean, I don’t wish to tell you how to run your website, but suppose you added something that grabbed a person’s attention? I mean BLOG_TITLE is kinda vanilla. You should look at Yahoo’s front page and watch how they create post headlines to grab viewers to click. You might add a video or a related picture or two to get people interested about what you’ve got to say. In my opinion, it could make your posts a little bit more interesting.
Hello to every body, it’s my first go to see of this website; this webpage consists of remarkable and truly excellent stuff designed for readers.
Its like you read my mind! You seem to know a lot about this, like you wrote the book in it or something. I think that you can do with some pics to drive the message home a bit, but instead of that, this is excellent blog. An excellent read. I’ll definitely be back.
Статья содержит подробное описание событий и контекста, при этом не выражая пристрастие к какой-либо стороне.
Post writing is also a fun, if you know then you can write if not it is complex to write.
Мне понравилась объективность и сбалансированность в подаче материала в статье.
Hmm is anyone else experiencing problems with the pictures on this blog loading? I’m trying to find out if its a problem on my end or if it’s the blog. Any suggestions would be greatly appreciated.
Автор предлагает реалистичные решения, которые могут быть внедрены в реальной жизни.
Читателям предоставляется возможность самостоятельно рассмотреть и проанализировать информацию.
Надеюсь, что эти комментарии добавят ещё больше позитива и поддержки к информационной статье! Это сообщение отправлено с сайта GoToTop.ee
Hello there! This is my first visit to your blog! We are a team of volunteers and starting a new initiative in a community in the same niche. Your blog provided us beneficial information to work on. You have done a wonderful job!
Статья предлагает глубокий анализ проблемы, рассматривая ее со всех сторон.
Автор старается сохранить нейтральность и оставляет решение оценки информации читателям.
Я хотел бы поблагодарить автора этой статьи за его основательное исследование и глубокий анализ. Он представил информацию с обширной перспективой и помог мне увидеть рассматриваемую тему с новой стороны. Очень впечатляюще!
Автор предлагает практические рекомендации, которые могут быть полезны в реальной жизни для решения проблемы.
Я хотел бы выразить признательность автору этой статьи за его объективный подход к теме. Он представил разные точки зрения и аргументы, что позволило мне получить полное представление о рассматриваемой проблеме. Очень впечатляюще!
Статья содержит актуальную статистику, что помогает оценить масштаб проблемы.
Автор старается сохранить нейтральность и предоставить балансированную информацию.
Heya i am for the first time here. I found this board and I find It really useful & it helped me out a lot. I hope to give something back and help others like you helped me.
Мне понравилось разнообразие источников, использованных автором для подкрепления своих утверждений.
I know this if off topic but I’m looking into starting my own blog and was wondering what all is required to get setup? I’m assuming having a blog like yours would cost a pretty penny? I’m not very internet savvy so I’m not 100 positive. Any suggestions or advice would be greatly appreciated. Appreciate it
Это способствует более глубокому пониманию темы и формированию информированного мнения.
Очень хорошо структурированная статья! Я оцениваю ясность и последовательность изложения. Благодаря этому, я смог легко следовать за логикой и усвоить представленную информацию. Большое спасибо автору за такой удобный формат!
Hi there every one, here every person is sharing such knowledge, therefore it’s fastidious to read this webpage, and I used to visit this website all the time.
Эта статья – настоящая находка! Она не только содержит обширную информацию, но и организована в простой и логичной структуре. Я благодарен автору за его усилия в создании такого интересного и полезного материала.
Автор представляет сложные понятия в доступной форме, что помогает лучше понять тему.
Я просто восхищен этой статьей! Автор предоставил глубокий анализ темы и подкрепил его примерами и исследованиями. Это помогло мне лучше понять предмет и расширить свои знания. Браво!
Мне понравилась четкая и логическая структура статьи, которая облегчает чтение.
Статья содержит информацию, подкрепленную примерами и исследованиями.
magnificent put up, very informative. I wonder why the other experts of this sector don’t notice this. You should continue your writing. I am sure, you have a great readers’ base already!
Мне понравился баланс между фактами и мнениями в статье.
Hi there I am so happy I found your website, I really found you by accident, while I was looking on Aol for something else, Regardless I am here now and would just like to say many thanks for a remarkable post and a all round thrilling blog (I also love the theme/design), I don’t have time to go through it all at the moment but I have saved it and also added in your RSS feeds, so when I have time I will be back to read much more, Please do keep up the fantastic work.
This is a topic that is near to my heart… Take care! Where are your contact details though?
Статья представляет анализ различных точек зрения на проблему.
Мне понравилась четкая логика аргументации в статье.
Автор старается оставаться нейтральным, предоставляя информацию для дальнейшего изучения.
Автор старается сохранить нейтральность, чтобы читатели могли основываться на объективной информации при формировании своего мнения. Это сообщение отправлено с сайта https://ru.gototop.ee/
Статья содержит дополнительные примеры, которые помогают проиллюстрировать основные концепции.
Good response in return of this difficulty with real arguments and explaining the whole thing on the topic of that.
You actually make it appear so easy along with your presentation but I to find this matter to be really something which I think I would never understand. It seems too complicated and very large for me. I’m looking forward for your next post, I’ll try to get the grasp of it!
Автор представляет разнообразные точки зрения на проблему, что помогает читателю получить обширное представление о ней.
Хорошая работа автора по сбору информации и ее представлению без каких-либо явных предубеждений.
Hi there Dear, are you actually visiting this website daily, if so after that you will without doubt obtain fastidious know-how.
Я очень доволен, что прочитал эту статью. Она оказалась настоящим открытием для меня. Информация была представлена в увлекательной и понятной форме, и я получил много новых знаний. Спасибо автору за такое удивительное чтение!
Автор старается представить материал нейтрально, что способствует обоснованному рассмотрению темы.
Это позволяет читателям самостоятельно сделать выводы и продолжить исследование по данному вопросу.
Статья ясно описывает факты и события, связанные с обсуждаемой темой.
I’m gone to tell my little brother, that he should also pay a quick visit this web site on regular basis to take updated from newest gossip.
Автор представляет сложные понятия в доступной форме, что помогает лучше понять тему.
Woah! I’m really enjoying the template/theme of this website. It’s simple, yet effective. A lot of times it’s very difficult to get that “perfect balance” between superb usability and visual appearance. I must say you have done a excellent job with this. In addition, the blog loads very quick for me on Firefox. Exceptional Blog!
Автор старается оставаться нейтральным, чтобы читатели могли самостоятельно рассмотреть различные точки зрения и сформировать собственное мнение. Это сообщение отправлено с сайта https://ru.gototop.ee/
Hello this is somewhat of off topic but I was wondering if blogs use WYSIWYG editors or if you have to manually code with HTML. I’m starting a blog soon but have no coding experience so I wanted to get advice from someone with experience. Any help would be greatly appreciated!
Nice blog right here! Additionally your website quite a bit up very fast! What web host are you the usage of? Can I get your associate hyperlink on your host? I desire my site loaded up as fast as yours lol
Надеюсь, что эти комментарии добавят ещё больше положительных настроений к информационной статье!
Автор старается подойти к теме нейтрально, чтобы предоставить читателям полную картину.
Hi I am so excited I found your site, I really found you by accident, while I was looking on Digg for something else, Anyhow I am here now and would just like to say thank you for a incredible post and a all round interesting blog (I also love the theme/design), I don’t have time to read it all at the minute but I have saved it and also included your RSS feeds, so when I have time I will be back to read more, Please do keep up the superb work.
This paragraph presents clear idea designed for the new viewers of blogging, that in fact how to do blogging.
Я прочитал эту статью с большим удовольствием! Она написана ясно и доступно, несмотря на сложность темы. Большое спасибо автору за то, что делает сложные понятия понятными для всех.
Hi there! I just want to give you a big thumbs up for your excellent info you have here on this post. I will be coming back to your blog for more soon.
Aw, this was an exceptionally good post. Taking a few minutes and actual effort to make a good article… but what can I say… I put things off a lot and don’t seem to get nearly anything done.
Hello, yeah this paragraph is truly nice and I have learned lot of things from it concerning blogging. thanks.
I will right away clutch your rss as I can not in finding your e-mail subscription link or e-newsletter service. Do you have any? Kindly permit me recognize in order that I could subscribe. Thanks.
Надеюсь, что эти комментарии добавят ещё больше положительных настроений к информационной статье!
Статья помогла мне лучше понять взаимосвязи между разными аспектами темы.
Aw, this was a really nice post. Finding the time and actual effort to make a really good article… but what can I say… I procrastinate a lot and never manage to get nearly anything done.
Я оцениваю степень детализации информации в статье, которая позволяет получить полное представление о проблеме.
Автор старается представить материал нейтрально, что помогает читателям обрести полное понимание обсуждаемой темы.
Good way of describing, and nice post to take information concerning my presentation subject matter, which i am going to deliver in academy.
Статья содержит анализ плюсов и минусов разных решений, связанных с проблемой.
Позиция автора не является однозначной, что позволяет читателям более глубоко разобраться в обсуждаемой теме.
Я только что прочитал эту статью, и мне действительно понравилось, как она написана. Автор использовал простой и понятный язык, несмотря на тему, и представил информацию с большой ясностью. Очень вдохновляюще!
Я оцениваю тщательность и точность исследования, представленного в этой статье. Автор провел глубокий анализ и представил аргументированные выводы. Очень важная и полезная работа!
Appreciate the recommendation. Will try it out.
Охранно-Защитных Дератизационных Систем (ОЗДС) в Москве и Санкт-Петербурге. Поставка оборудования для ОЗДС по всей России. Проект. Монтаж. Обслуживание.
Установка Охранно-Защитной Дератизационной Системы требует профессиональных навыков и опыта, чтобы не нанести вреда остальным живым существам и обеспечить безопасность людей, находящихся в помещении. Поэтому важно обратиться к специалистам с соответствующей лицензией и сертификатами на проведение дератизации. Охранно-защитная дератизационная система – надежный метод защиты от грызунов в любых условиях. Для достижения наилучшего результата следует обратиться к профессионалам, которые обеспечат качественное и безопасное проведение дератизации.
Its like you read my mind! You appear to know so much about this, like you wrote the book in it or something. I think that you could do with a few pics to drive the message home a little bit, but other than that, this is great blog. A great read. I’ll certainly be back.
Fastidious answers in return of this difficulty with real arguments and telling the whole thing on the topic of that.
Great blog! Is your theme custom made or did you download it from somewhere? A design like yours with a few simple tweeks would really make my blog jump out. Please let me know where you got your theme. Appreciate it
Wow! This blog looks exactly like my old one! It’s on a completely different topic but it has pretty much the same layout and design. Outstanding choice of colors!
I’ve learn some good stuff here. Definitely price bookmarking for revisiting. I surprise how so much effort you place to make one of these great informative site.
I’ve been surfing online more than three hours today, yet I never found any interesting article like yours. It’s pretty worth enough for me. In my view, if all web owners and bloggers made good content as you did, the web will be much more useful than ever before.
Helpful info. Fortunate me I discovered your site by chance, and I am surprised why this accident didn’t came about in advance! I bookmarked it.
Hey There. I found your weblog using msn. That is an extremely smartly written article. I will make sure to bookmark it and come back to read more of your helpful information. Thanks for the post. I’ll definitely comeback.
You could certainly see your expertise within the work you write. The sector hopes for more passionate writers such as you who aren’t afraid to say how they believe. Always follow your heart.
Great post. I used to be checking constantly this weblog and I’m impressed! Extremely useful information particularly the closing part 🙂 I handle such info much. I was looking for this particular info for a long time. Thank you and best of luck.
I am really impressed together with your writing abilities and also with the layout in your weblog. Is that this a paid topic or did you customize it your self? Either way stay up the nice quality writing, it is uncommon to look a nice blog like this one nowadays..
Hmm is anyone else having problems with the pictures on this blog loading? I’m trying to find out if its a problem on my end or if it’s the blog. Any feedback would be greatly appreciated.
It’s very simple to find out any topic on net as compared to books, as I found this paragraph at this web page.
My brother suggested I might like this web site. He used to be totally right. This publish actually made my day. You can not consider simply how a lot time I had spent for this info! Thank you!
What’s Happening i am new to this, I stumbled upon this I have discovered It absolutely helpful and it has helped me out loads. I am hoping to contribute & aid different customers like its helped me. Good job.
Hi, I check your blog like every week. Your writing style is witty, keep doing what you’re doing!
Saved as a favorite, I love your website!
Oh my goodness! Amazing article dude! Thank you so much, However I am experiencing troubles with your RSS. I don’t know the reason why I cannot subscribe to it. Is there anybody having identical RSS issues? Anybody who knows the solution can you kindly respond? Thanx!!
Howdy, There’s no doubt that your website may be having internet browser compatibility problems. Whenever I look at your blog in Safari, it looks fine however, if opening in I.E., it’s got some overlapping issues. I just wanted to provide you with a quick heads up! Other than that, fantastic blog!
If you would like to grow your knowledge only keep visiting this site and be updated with the most recent news posted here.
Very soon this website will be famous amid all blog users, due to it’s good content
Do you have a spam issue on this site; I also am a blogger, and I was wondering your situation; many of us have developed some nice methods and we are looking to exchange strategies with other folks, please shoot me an e-mail if interested.
Good day! Do you know if they make any plugins to help with Search Engine Optimization? I’m trying to get my blog to rank for some targeted keywords but I’m not seeing very good results. If you know of any please share. Cheers!
I seriously love your website.. Great colors & theme. Did you create this site yourself? Please reply back as I’m trying to create my very own blog and want to learn where you got this from or just what the theme is named. Kudos!
I was wondering if you ever considered changing the page layout of your blog? Its very well written; I love what youve got to say. But maybe you could a little more in the way of content so people could connect with it better. Youve got an awful lot of text for only having 1 or 2 pictures. Maybe you could space it out better?
Hurrah! Finally I got a web site from where I can actually take helpful information concerning my study and knowledge.
Since the admin of this site is working, no uncertainty very shortly it will be famous, due to its feature contents.
Wonderful site. A lot of useful info here. I am sending it to some friends ans also sharing in delicious. And of course, thanks for your effort!
What’s up everyone, it’s my first go to see at this web page, and post is genuinely fruitful designed for me, keep up posting these content.
Hi, after reading this awesome piece of writing i am too glad to share my experience here with mates.
Thanks for the good writeup. It if truth be told was once a entertainment account it. Look complex to more introduced agreeable from you! By the way, how could we be in contact?
After I initially commented I seem to have clicked on the -Notify me when new comments are added- checkbox and from now on every time a comment is added I receive 4 emails with the exact same comment. Is there a means you are able to remove me from that service? Many thanks!
Thanks , I’ve just been looking for information about this topic for ages and yours is the greatest I have found out so far. However, what concerning the conclusion? Are you positive in regards to the source?
Appreciate the recommendation. Will try it out.
Heya i’m for the first time here. I found this board and I find It really helpful & it helped me out a lot. I am hoping to provide one thing again and aid others such as you helped me.
Can I simply say what a relief to uncover somebody that truly knows what they are discussing online. You definitely know how to bring an issue to light and make it important. More people need to look at this and understand this side of your story. It’s surprising you aren’t more popular since you most certainly have the gift.
Great blog! Do you have any tips for aspiring writers? I’m planning to start my own website soon but I’m a little lost on everything. Would you recommend starting with a free platform like WordPress or go for a paid option? There are so many options out there that I’m completely overwhelmed .. Any tips? Thanks!
Thanks for finally talking about > blog_title < Liked it!
I have been exploring for a little bit for any high-quality articles or weblog posts in this sort of house . Exploring in Yahoo I at last stumbled upon this site. Reading this info So i’m satisfied to express that I have an incredibly excellent uncanny feeling I came upon exactly what I needed. I most surely will make sure to don?t put out of your mind this web site and give it a glance regularly.
Hey I know this is off topic but I was wondering if you knew of any widgets I could add to my blog that automatically tweet my newest twitter updates. I’ve been looking for a plug-in like this for quite some time and was hoping maybe you would have some experience with something like this. Please let me know if you run into anything. I truly enjoy reading your blog and I look forward to your new updates.
Hello There. I found your blog using msn. This is a very well written article. I’ll be sure to bookmark it and come back to read more of your useful info. Thanks for the post. I will definitely comeback.
I like the helpful information you supply for your articles. I’ll bookmark your blog and check once more here frequently. I’m somewhat certain I’ll be informed lots of new stuff right here! Best of luck for the following!
Pretty element of content. I just stumbled upon your site and in accession capital to claim that I get in fact loved account your weblog posts. Anyway I will be subscribing for your augment and even I fulfillment you get admission to consistently fast.
I’m not that much of a online reader to be honest but your blogs really nice, keep it up! I’ll go ahead and bookmark your website to come back later. Cheers
Thanks for the marvelous posting! I certainly enjoyed reading it, you’re a great author.I will be sure to bookmark your blog and may come back later on. I want to encourage you continue your great writing, have a nice day!
Ahaa, its nice conversation about this post here at this web site, I have read all that, so at this time me also commenting here.
I used to be recommended this website by means of my cousin. I’m no longer positive whether this publish is written via him as no one else recognize such specific about my trouble. You’re incredible! Thanks!
I always spent my half an hour to read this web site’s content everyday along with a cup of coffee.
You made some decent points there. I checked on the net for more info about the issue and found most people will go along with your views on this site.
I’m curious to find out what blog platform you’re working with? I’m having some minor security problems with my latest site and I’d like to find something more safe. Do you have any solutions?
Hello! This is my first visit to your blog! We are a collection of volunteers and starting a new project in a community in the same niche. Your blog provided us useful information to work on. You have done a wonderful job!
Somebody essentially lend a hand to make seriously posts I might state. This is the very first time I frequented your web page and to this point? I surprised with the analysis you made to make this actual submit amazing. Excellent activity!
You are so interesting! I don’t think I’ve read something like that before. So wonderful to find someone with some unique thoughts on this issue. Seriously.. thank you for starting this up. This web site is something that is needed on the internet, someone with some originality!
Undeniably believe that which you said. Your favorite justification appeared to be on the web the easiest thing to be aware of. I say to you, I definitely get irked while people consider worries that they just do not know about. You managed to hit the nail upon the top and also defined out the whole thing without having side effect , people could take a signal. Will likely be back to get more. Thanks
Hi, I think your site might be having browser compatibility issues. When I look at your blog in Opera, it looks fine but when opening in Internet Explorer, it has some overlapping. I just wanted to give you a quick heads up! Other then that, superb blog!
I always spent my half an hour to read this website’s content every day along with a cup of coffee.
I used to be suggested this website by my cousin. I am no longer sure whether this publish is written by means of him as nobody else know such precise about my difficulty. You’re amazing! Thanks!
I’m very pleased to find this site. I want to to thank you for your time for this particularly fantastic read!! I definitely appreciated every little bit of it and I have you bookmarked to see new information in your website.
Thanks for sharing your thoughts about meta_keyword. Regards
What a data of un-ambiguity and preserveness of valuable know-how about unexpected emotions.
I enjoy what you guys are up too. This kind of clever work and exposure! Keep up the good works guys I’ve included you guys to blogroll.
Greate pieces. Keep writing such kind of information on your page. Im really impressed by it.
I am extremely impressed along with your writing abilities as well as with the format to your blog. Is that this a paid topic or did you customize it your self? Either way stay up the nice quality writing, it’s uncommon to see a great blog like this one today..
Yesterday, while I was at work, my cousin stole my iPad and tested to see if it can survive a thirty foot drop, just so she can be a youtube sensation. My iPad is now destroyed and she has 83 views. I know this is entirely off topic but I had to share it with someone!
Thanks very nice blog!
You need to take part in a contest for one of the greatest sites online. I’m going to recommend this website!
Hello, its good post about media print, we all be aware of media is a impressive source of information.
Hello! I just wanted to ask if you ever have any trouble with hackers? My last blog (wordpress) was hacked and I ended up losing months of hard work due to no data backup. Do you have any methods to prevent hackers?
If you are going for most excellent contents like me, only go to see this site every day since it presents quality contents, thanks
Yes! Finally something about keyword1.
bookmarked!!, I like your site!
Статья помогла мне лучше понять сложные взаимосвязи в данной теме.
It’s fantastic that you are getting thoughts from this post as well as from our dialogue made at this time.
Статья помогла мне лучше понять взаимосвязи между разными аспектами темы.
Я не могу не отметить качество исследования, представленного в этой статье. Она обогатила мои знания и вдохновила меня на дальнейшее изучение темы. Благодарю автора за его ценный вклад!
Статья предлагает читателям объективную информацию, подкрепленную проверенными источниками.
Я оцениваю объективность и сбалансированность аргументации в статье.
I’m really impressed with your writing skills and also with the layout on your blog. Is this a paid theme or did you modify it yourself? Anyway keep up the excellent quality writing, it is rare to see a great blog like this one these days.
Hey there! Someone in my Facebook group shared this site with us so I came to look it over. I’m definitely loving the information. I’m bookmarking and will be tweeting this to my followers! Excellent blog and wonderful style and design.
Я прочитал эту статью с огромным интересом! Автор умело объединил факты, статистику и персональные истории, что делает ее настоящей находкой. Я получил много новых знаний и вдохновения. Браво!
Статья содержит актуальную статистику, что помогает оценить масштаб проблемы.
Хорошая работа по анализу проблемы и представлению различных точек зрения.
Читатели имеют возможность самостоятельно проанализировать представленные факты и сделать собственные выводы.
Автор умело структурирует информацию, что помогает сохранить интерес читателя.
It is not my first time to pay a quick visit this web page, i am browsing this site dailly and obtain fastidious data from here every day.
I’m not that much of a internet reader to be honest but your sites really nice, keep it up! I’ll go ahead and bookmark your website to come back down the road. Cheers
Автор не вмешивается в читателей, а предоставляет им возможность самостоятельно оценить представленную информацию.
Thanks a lot for sharing this with all folks you actually recognise what you are talking approximately! Bookmarked. Please also consult with my site =). We could have a hyperlink change arrangement among us
I’ve been surfing online more than 2 hours today, yet I never found any interesting article like yours. It is pretty worth enough for me. In my view, if all website owners and bloggers made good content as you did, the net will be much more useful than ever before.
Я оцениваю объективный подход автора и его стремление представить полную картину проблемы.
Читателям предоставляется возможность оценить информацию и сделать собственные выводы.
Автор старается представить материал нейтрально, что способствует обоснованному рассмотрению темы.
Мне понравился стиль изложения в статье, который делает ее легко читаемой и понятной.
Мне понравилась четкая и последовательная логика аргументации автора в статье.
What’s up mates, its fantastic post concerning tutoringand completely defined, keep it up all the time.
Одним из важных аспектов ОЗДС является также обучение и информирование персонала, работающего в зданиях и помещениях, о мерах предотвращения проникновения грызунов. Персонал должен знать, как распознавать признаки наличия грызунов, как правильно обращаться с мусором, продуктами питания и другими материалами, и как реагировать в случае обнаружения грызунов или их следов. Регулярное обучение и информирование персонала позволяют повысить осведомленность и ответственность в отношении ОЗДС и содействуют эффективной защите от грызунов.
Охранно-защитные дератизационные системы (ОЗДС) – это эффективный способ избавления от грызунов на территории практически любого объекта недвижимости и предотвращения их проникновения извне путем создания электрического барьера. ОЗДС работает на основе электрического заряда, который создает барьер для грызунов. Грызуны не могут перепрыгнуть через барьер и не могут пройти через него.
Одним из ключевых компонентов ОЗДС является система мониторинга и предотвращения проникновения грызунов. Эта система может включать в себя различные средства, такие как запирательные устройства, преграды, ловушки, а также мониторинговое оборудование, позволяющее отслеживать движение грызунов и своевременно реагировать на их присутствие. Также может быть применена автоматическая система оповещения, которая мгновенно информирует операторов о нарушениях и позволяет принять меры по их предотвращению.
Охранно-Защитная Дератизационная Система (ОЗДС) представляет собой комплекс мер и технических средств, предназначенных для борьбы с грызунами. Она используется для защиты от вредоносных грызунов, таких как крысы и мыши, которые могут нанести ущерб зданиям, техническим коммуникациям, продуктам питания и здоровью людей.
Для борьбы с грызунами в ОЗДС используются различные методы, включая применение специализированных препаратов, ловушек и устройств с физическим воздействием на грызунов. Важной особенностью ОЗДС является минимальное использование ядовитых веществ, что делает систему экологически безопасной и безопасной для людей и домашних животных.
Одной из ключевых особенностей ОЗДС является ее комплексный подход к борьбе с грызунами. Система предлагает несколько методов и технологий, таких как электромагнитные устройства, ультразвуковые излучатели, световые и звуковые отпугиватели, физические барьеры и другие. Такой разнообразный подход позволяет эффективно противодействовать грызунам различных видов, таким как крысы, мыши, белки и другие, и предотвращать их проникновение на объект.
Охранно-Защитная Дератизационная Система (ОЗДС) использует электротехнический комплекс для эффективной защиты помещений и различных коммуникаций здания от грызунов, мышей и крыс. ОЗДС работает путем перекрытия подходов к местам кормления, локализации возможных мест гнездования и перекрытия путей их постоянных перемещений. Технология ОЗДС включает генератор электрических импульсных разрядов, который электрически связан с одним или несколькими электризуемыми барьерами³. Это позволяет создать электрический барьер для грызунов и предотвратить их проникновение в помещение.
ОЗДС также может включать применение химических средств борьбы с грызунами, таких как отравы и репелленты. Однако, важно отметить, что применение химических средств должно быть произведено квалифицированными специалистами с соблюдением всех необходимых мер предосторожности и в соответствии с действующим законодательством. Химические средства должны быть использованы в строгом соответствии с инструкциями производителя и только в тех местах, где они безопасны для людей и домашних животных.
Одним из важных аспектов ОЗДС является также обучение и информирование персонала, работающего в зданиях и помещениях, о мерах предотвращения проникновения грызунов.
Помимо мер предотвращения, ОЗДС также включает мониторинг и контроль за наличием грызунов в зданиях и помещениях. Это может включать регулярные осмотры, анализ следов, а также мониторинговые системы, которые позволяют оперативно обнаруживать наличие грызунов и принимать меры по их эффективному устранению.
ОЗДС также предлагает разнообразные методы дератизации, адаптирующиеся под специфику объекта и вид грызунов. Это может включать применение гелевых ловушек, ультразвуковых отпугивателей, электромагнитных блокировщиков, инфразвуковых генераторов, автоматических дозаторов яда и других современных технологий. Одновременное использование нескольких методов дератизации обеспечивает максимальную эффективность и снижает возможность адаптации грызунов к одному методу.
ОЗДС является важной составляющей программы санитарной обработки территорий и помещений, и она помогает предотвратить распространение заболеваний, передаваемых грызунами, и уменьшить материальные убытки, связанные с их активностью. Для эффективной реализации ОЗДС рекомендуется обращаться к профессионалам, имеющим соответствующую квалификацию и опыт в области дератизации.
Охранно-Защитная Дератизационная Система (ОЗДС) представляет собой комплекс мер и технических средств, предназначенных для борьбы с грызунами. Она используется для защиты от вредоносных грызунов, таких как крысы и мыши, которые могут нанести ущерб зданиям, техническим коммуникациям, продуктам питания и здоровью людей.
Я хотел бы поблагодарить автора этой статьи за его основательное исследование и глубокий анализ. Он представил информацию с обширной перспективой и помог мне увидеть рассматриваемую тему с новой стороны. Очень впечатляюще!
Я оцениваю объективный и непредвзятый подход автора к теме.
Я прочитал эту статью с большим удовольствием! Автор умело смешал факты и личные наблюдения, что придало ей уникальный характер. Я узнал много интересного и наслаждался каждым абзацем. Браво!
Heya i am for the primary time here. I found this board and I to find It really useful & it helped me out a lot. I’m hoping to provide one thing again and aid others such as you aided me.
Marvelous, what a webpage it is! This website presents valuable facts to us, keep it up.
I savor, lead to I discovered just what I was looking for. You have ended my four day lengthy hunt! God Bless you man. Have a great day. Bye
Я хотел бы выразить свою восторженность этой статьей! Она не только информативна, но и вдохновляет меня на дальнейшее изучение темы. Автор сумел передать свою страсть и знания, что делает эту статью поистине уникальной.
Автор предлагает реалистичные решения, которые могут быть внедрены в реальной жизни.
This is very interesting, You are a very skilled blogger. I’ve joined your rss feed and look forward to seeking more of your fantastic post. Also, I have shared your site in my social networks!
Я чувствую, что эта статья является настоящим источником вдохновения. Она предлагает новые идеи и вызывает желание узнать больше. Большое спасибо автору за его творческий и информативный подход!
Я ценю фактический и информативный характер этой статьи. Она предлагает читателю возможность рассмотреть различные аспекты рассматриваемой проблемы без внушения какого-либо определенного мнения.
Существует несколько видов дератизации, которые позволяют бороться с различными видами грызунов. 1. Механическая дератизация – это один из самых простых и доступных способов борьбы с грызунами, который основывается на физическом уничтожении грызунов и их гнезд. Для механической дератизации используются ловушки, клейкие ловушки, сетки и другие приспособления. 2. Химическая дератизация – это метод, который основывается на применении ядовитых препаратов для уничтожения грызунов. Для химической дератизации используются различные ядовитые препараты, которые выпускаются в виде локальных препаратов, растворов и аэрозолей. 3. Биологическая дератизация – это метод, который основывается на использовании естественных врагов грызунов. В качестве естественных врагов используются крысы, ястребы, бурундуки и другие животные. В зависимости от результата, который необходимо достичь, выбирается соответствующий вид дератизации. При этом, необходимо учитывать все моменты, связанные с безопасностью работы и возможными последствиями.
Статья дает полное представление об Охранно-Защитной Дератизационной Системе (ОЗДС) и ее роли в обеспечении безопасности от грызунов. ОЗДС предлагает разнообразные методы и стратегии, включая превентивные меры, мониторинг и уничтожение грызунов. Статья акцентирует внимание на преимуществах ОЗДС, таких как предотвращение повреждений имущества, снижение рисков для здоровья и контроль численности грызунов. Эта система играет важную роль в обеспечении безопасности в различных сферах, от домашнего использования до коммерческих и промышленных объектов. Регулярное обслуживание и обучение персонала по использованию ОЗДС являются ключевыми факторами для эффективности системы.
Статья очень информативна по поводу Охранно-Защитной Дератизационной Системы (ОЗДС). Она подробно описывает принципы работы системы и ее важность в борьбе с грызунами и обеспечении безопасности. ОЗДС предлагает широкий спектр методов, включая применение химических средств, ловушек и электронных систем мониторинга. Это помогает предотвратить повреждения имущества, контролировать популяцию грызунов и снизить риск распространения болезней. Статья также обращает внимание на важность регулярного обслуживания и обучения персонала для оптимальной работы ОЗДС. В целом, ОЗДС играет ключевую роль в создании безопасной и гигиеничной среды, и эта статья отлично передает ее значимость.
Статья подробно описывает Охранно-Защитную Дератизационную Систему (ОЗДС) и ее важность в обеспечении безопасности от грызунов. ОЗДС предлагает интегрированный подход, который включает в себя предотвращение, обнаружение и уничтожение грызунов. Статья указывает на разнообразие методов и технологий, доступных в рамках ОЗДС, таких как применение ядовитых препаратов, ловушек, электронных систем мониторинга и физических барьеров. ОЗДС является неотъемлемой частью программы по обеспечению безопасности и гигиены, и ее применение позволяет предотвратить повреждения имущества, риски для здоровья и распространение заболеваний, переносимых грызунами.
Статья о Охранно-Защитной Дератизационной Системе (ОЗДС) привлекает внимание к важности этой системы в борьбе с грызунами и обеспечении безопасности. ОЗДС предлагает разнообразные методы, включая использование экологически безопасных препаратов, ловушек и электронных систем мониторинга. Это позволяет эффективно контролировать грызунов и предотвращать их проникновение, минимизируя риски для здоровья и предотвращая повреждения имущества. Статья также подчеркивает важность регулярного обслуживания и обучения персонала, чтобы гарантировать надежность и эффективность ОЗДС. ОЗДС является неотъемлемой частью программы по обеспечению безопасности в различных сферах, и эта статья отлично передает ее важность.
Барьерный элемент системы ОЗДС устанавливают в различных местах, в зависимости от целей использования системы и конкретных условий объекта, на который она устанавливается. Одним из основных мест, где устанавливают барьерный элемент системы ОЗДС, являются зоны периметра объекта. В таких местах барьер обеспечивает контроль и ограничение доступа на территорию объекта. Он может быть установлен на ограждения, стены, заборы, калитки и другие объекты, обеспечивающие безопасность и защиту периметра. Барьер может быть как непроницаемым, так и иметь различные степени проницаемости, в зависимости от режима охраны объекта.
ОЗДС является неотъемлемым компонентом программы санитарного обслуживания и обеспечивает высокую степень защиты от грызунов. Эта система предлагает широкий спектр методов и средств, включая применение препаратов, установку ловушек и блокирование доступа грызунов. ОЗДС также включает в себя регулярные инспекции и аудиты, чтобы гарантировать эффективность системы на протяжении времени. Она играет важную роль в поддержании чистоты, гигиены и безопасности в различных сферах, от производственных помещений до жилых зон.
I visited many web sites but the audio feature for audio songs present at this website is really marvelous.
Очень хорошо структурированная статья! Я оцениваю ясность и последовательность изложения. Благодаря этому, я смог легко следовать за логикой и усвоить представленную информацию. Большое спасибо автору за такой удобный формат!
Радует объективность статьи, автор старается представить информацию без сильной эмоциональной окраски.
Статья содержит подробное описание событий и контекста, при этом не выражая пристрастие к какой-либо стороне.
Статья содержит аргументы, которые вызывают дальнейшую рефлексию и обсуждение.
Я оцениваю тщательность и точность, с которыми автор подошел к составлению этой статьи. Он привел надежные источники и представил информацию без преувеличений. Благодаря этому, я могу доверять ей как надежному источнику знаний.
Статья помогла мне лучше понять сложные аспекты темы, с которыми я ранее не сталкивался.
Надеюсь, что эти дополнительные комментарии принесут ещё больше позитивных отзывов на информационную статью! Это сообщение отправлено с сайта GoToTop.ee
Я ценю балансировку автора в описании проблемы. Он предлагает читателю достаточно аргументов и контекста для формирования собственного мнения, не внушая определенную точку зрения.
Отличная статья! Я бы хотел отметить ясность и логичность, с которыми автор представил информацию. Это помогло мне легко понять сложные концепции. Большое спасибо за столь прекрасную работу!
Автор предлагает практические рекомендации, основанные на исследованиях и опыте.
Автор предлагает систематический анализ проблемы, учитывая разные точки зрения.
Я хотел бы выразить свою восторженность этой статьей! Она не только информативна, но и вдохновляет меня на дальнейшее изучение темы. Автор сумел передать свою страсть и знания, что делает эту статью поистине уникальной.
Позиция автора остается нейтральной, что позволяет читателям сформировать свое мнение.
Автор статьи предлагает разные точки зрения на обсуждаемую проблему, позволяя читателям ознакомиться с различными аргументами и сделать собственные выводы.
Автор предлагает обоснованные и логические выводы на основе представленных фактов и данных.
Автор статьи представляет сведения, опираясь на факты и экспертные мнения.
Я просто не могу пройти мимо этой статьи без оставления положительного комментария. Она является настоящим примером качественной журналистики и глубокого исследования. Очень впечатляюще!
Статья содержит разнообразные точки зрения, представленные в равной мере.
Автор предоставляет подробные сведения и контекст, что помогает читателям лучше понять обсуждаемую тему.
Я благодарен автору этой статьи за его тщательное и глубокое исследование. Он представил информацию с большой детализацией и аргументацией, что делает эту статью надежным источником знаний. Очень впечатляющая работа!
Thanks for sharing your info. I really appreciate your efforts and I will be waiting for your next write ups thanks once again.
Статья помогла мне получить более полное представление о проблеме, которая рассматривается.
Статья содержит анализ преимуществ и недостатков различных решений, связанных с темой.
Мне понравилась глубина исследования, представленная в статье.
You have made some decent points there. I checked on the internet for more information about the issue and found most individuals will go along with your views on this web site.
What’s Going down i’m new to this, I stumbled upon this I have found It absolutely useful and it has aided me out loads. I hope to give a contribution & aid different customers like its aided me. Good job.
Хорошо, что автор обратил внимание на различные аспекты данной проблемы.
Я хотел бы выразить признательность автору за его глубокое понимание темы и его способность представить информацию во всей ее полноте. Я по-настоящему насладился этой статьей и узнал много нового!
Статья содержит анализ причин и последствий проблемы, что позволяет лучше понять ее важность и сложность.
Автор старается оставаться объективным, чтобы читатели могли сформировать свое собственное мнение на основе предоставленной информации.
I truly love your blog.. Great colors & theme. Did you make this amazing site yourself? Please reply back as I’m attempting to create my own personal blog and would like to learn where you got this from or just what the theme is named. Thank you!
It’s nearly impossible to find well-informed people for this subject, but you seem like you know what you’re talking about! Thanks
Статья помогла мне лучше понять взаимосвязи между разными аспектами темы.
I know this web site offers quality dependent content and additional information, is there any other web page which presents these stuff in quality?
Thanks for ones marvelous posting! I actually enjoyed reading it, you happen to be a great author.I will always bookmark your blog and definitely will come back at some point. I want to encourage yourself to continue your great job, have a nice afternoon!
Hello to all, how is all, I think every one is getting more from this web site, and your views are nice designed for new visitors.
Автор старается сохранить нейтральность, предоставляя обстоятельную основу для дальнейшего рассмотрения темы.
Надеюсь, что эти комментарии добавят ещё больше положительных настроений к информационной статье! Это сообщение отправлено с сайта GoToTop.ee
Автор предлагает дополнительные ресурсы, которые помогут читателю углубиться в тему и расширить свои знания.
Does your website have a contact page? I’m having trouble locating it but, I’d like to send you an e-mail. I’ve got some creative ideas for your blog you might be interested in hearing. Either way, great site and I look forward to seeing it grow over time.
Everything is very open with a clear description of the issues. It was truly informative. Your site is extremely helpful. Many thanks for sharing!
Hi mates, good paragraph and good urging commented at this place, I am truly enjoying by these.
Эта статья действительно отличная! Она предоставляет обширную информацию и очень хорошо структурирована. Я узнал много нового и интересного. Спасибо автору за такую информативную работу!
Статья содержит достаточно информации для того, чтобы читатель мог сделать собственные выводы.
Я хотел бы выразить свою благодарность автору за его глубокие исследования и ясное изложение. Он сумел объединить сложные концепции и представить их в доступной форме. Это действительно ценный ресурс для всех, кто интересуется этой темой.
I’m not that much of a internet reader to be honest but your sites really nice, keep it up! I’ll go ahead and bookmark your site to come back down the road. Cheers
Статья помогает читателю получить полное представление о проблеме, рассматривая ее с разных сторон.
I think the admin of this web site is really working hard in support of his web site, because here every material is quality based information.
Я только что прочитал эту статью, и мне действительно понравилось, как она написана. Автор использовал простой и понятный язык, несмотря на тему, и представил информацию с большой ясностью. Очень вдохновляюще!
Hello this is somewhat of off topic but I was wanting to know if blogs use WYSIWYG editors or if you have to manually code with HTML. I’m starting a blog soon but have no coding know-how so I wanted to get advice from someone with experience. Any help would be enormously appreciated!
Очень хорошо организованная статья! Автор умело структурировал информацию, что помогло мне легко следовать за ней. Я ценю его усилия в создании такого четкого и информативного материала.
Автор старается оставаться нейтральным, предоставляя информацию, не оказывающую явного влияния на читателей.
Статья представляет обширный обзор темы и учитывает ее исторический контекст.
Автор предоставляет анализ последствий проблемы и возможных путей ее решения.
Автор статьи предоставляет информацию, подкрепленную исследованиями и доказательствами, без выражения личных предпочтений. Это сообщение отправлено с сайта https://ru.gototop.ee/
I used to be able to find good info from your articles.
Читателям предоставляется возможность самостоятельно сформировать свое мнение на основе представленных фактов.
Мне понравилось разнообразие информации в статье, которое позволяет рассмотреть проблему с разных сторон.
Статья содержит дополнительные примеры, которые помогают проиллюстрировать основные концепции.
What’s up, I check your blog daily. Your story-telling style is awesome, keep doing what you’re doing!
Позиция автора остается нейтральной, что позволяет читателям сформировать свое мнение.
Статья помогла мне лучше понять сложные взаимосвязи в данной теме.
Мне понравилась аргументация автора, основанная на логической цепочке рассуждений.
There is certainly a great deal to find out about this subject. I really like all of the points you have made.
For hottest news you have to go to see the web and on world-wide-web I found this website as a best web site for most up-to-date updates.
Это позволяет читателям самостоятельно оценить и проанализировать информацию.
Автор предоставляет читателю возможность взглянуть на проблему с разных сторон.
Я оцениваю объективность автора и его стремление представить разные точки зрения на проблему.
Aw, this was an incredibly nice post. Finding the time and actual effort to create a good article… but what can I say… I procrastinate a lot and never manage to get anything done.
Читатели имеют возможность ознакомиться с разными точками зрения и самостоятельно оценить информацию.
Автор умело структурирует информацию, что помогает сохранить интерес читателя на протяжении всей статьи.
Эта статья – настоящая находка! Она не только содержит обширную информацию, но и организована в простой и логичной структуре. Я благодарен автору за его усилия в создании такого интересного и полезного материала.
Автор не старается убедить читателей в определенном мнении, а предоставляет информацию для самостоятельной оценки.
Автор старается оставаться нейтральным, позволяя читателям сами сформировать свое мнение на основе представленной информации.
Fantastic blog! Do you have any tips and hints for aspiring writers? I’m planning to start my own site soon but I’m a little lost on everything. Would you propose starting with a free platform like WordPress or go for a paid option? There are so many choices out there that I’m completely confused .. Any recommendations? Thanks a lot!
Это помогает читателям получить полную картину и сделать собственные выводы.
Это помогает читателям осознать сложность проблемы и самостоятельно сформировать свое собственное мнение.
Я не могу не отметить качество исследования, представленного в этой статье. Автор использовал надежные источники и предоставил нам актуальную информацию. Большое спасибо за такой надежный и информативный материал!
Hey there! This is kind of off topic but I need some help from an established blog. Is it difficult to set up your own blog? I’m not very techincal but I can figure things out pretty quick. I’m thinking about creating my own but I’m not sure where to start. Do you have any tips or suggestions? Appreciate it
Я бы хотел отметить качество исследования, проведенного автором этой статьи. Он представил обширный объем информации, подкрепленный надежными источниками. Очевидно, что автор проявил большую ответственность в подготовке этой работы.
Информационная статья представляет данные и факты, сопровождаемые объективным анализом.
Статья содержит актуальную информацию, которая помогает понять сложность и важность проблемы.
I enjoy reading an article that will make men and women think. Also, many thanks for permitting me to comment!
Статья предлагает глубокий анализ проблемы, рассматривая ее со всех сторон.
Автор старается быть нейтральным, чтобы читатели могли самостоятельно рассмотреть различные аспекты темы.
Эта статья – источник вдохновения и новых знаний! Я оцениваю уникальный подход автора и его способность представить информацию в увлекательной форме. Это действительно захватывающее чтение!
Мне понравилась объективность автора и его способность представить информацию без предвзятости.
В современном мире, где онлайн-присутствие становится все более важным для бизнеса, повышение видимости и ранжирования сайта в поисковых системах является одной из самых важных задач для веб-мастеров и маркетологов. Одним из основных факторов, влияющих на рост авторитетности сайта, является его DR (Domain Rating), который определяется в основном путем анализа ссылочной массы сайта.
I am genuinely thankful to the owner of this website who has shared this great piece of writing at here.
This text is priceless. Where can I find out more?
Автор представляет свои идеи объективно и не прибегает к эмоциональным уловкам.
Я оцениваю широту охвата темы в статье.
Статья помогла мне лучше понять контекст и значение проблемы в современном обществе.
Приятно видеть, что автор не делает однозначных выводов, а предоставляет читателям возможность самостоятельно анализировать представленные факты.
Статья предлагает разнообразные точки зрения на тему, предоставляя читателям возможность рассмотреть различные аспекты проблемы.
Автор предлагает читателю разные взгляды на проблему, что способствует формированию собственного мнения.
Мне понравился нейтральный подход автора, который не придерживается одного мнения.
Автор предоставляет актуальную информацию, которая помогает читателю быть в курсе последних событий и тенденций.
Статья помогла мне получить глубокое понимание проблемы, о которой я раньше не задумывался.
Way cool! Some very valid points! I appreciate you writing this article plus the rest of the website is extremely good.
Очень интересная исследовательская работа! Статья содержит актуальные факты, аргументированные доказательствами. Это отличный источник информации для всех, кто хочет поглубже изучить данную тему.
Я оцениваю тщательность и точность, с которыми автор подошел к составлению этой статьи. Он привел надежные источники и представил информацию без преувеличений. Благодаря этому, я могу доверять ей как надежному источнику знаний.
Автор представил четкую и структурированную статью, основанную на фактах и статистике.
I’m impressed, I must say. Seldom do I come across a blog that’s both educative and interesting, and let me tell you, you’ve hit the nail on the head. The problem is something that too few folks are speaking intelligently about. Now i’m very happy I came across this in my search for something relating to this.
It’s great that you are getting thoughts from this piece of writing as well as from our dialogue made at this place.
I’m impressed, I have to admit. Rarely do I come across a blog that’s equally educative and entertaining, and let me tell you, you have hit the nail on the head. The problem is an issue that not enough men and women are speaking intelligently about. I’m very happy that I found this during my hunt for something regarding this.
Автор старается не высказывать собственного мнения, что способствует нейтральному освещению темы.
Great info. Lucky me I came across your website by chance (stumbleupon). I have saved as a favorite for later!
Автор предоставляет читателю возможность взглянуть на проблему с разных сторон.
Статья представляет объективную оценку проблемы, учитывая мнение разных экспертов и специалистов.
Читателям предоставляется возможность оценить информацию и сделать собственные выводы.
Статья помогла мне лучше понять контекст и значение проблемы в современном обществе.
Hey! This post could not be written any better! Reading this post reminds me of my previous room mate! He always kept chatting about this. I will forward this article to him. Fairly certain he will have a good read. Thanks for sharing!
Very descriptive blog, I liked that bit. Will there be a part 2?
Эта статья превзошла мои ожидания! Она содержит обширную информацию, иллюстрирует примерами и предлагает практические советы. Я благодарен автору за его усилия в создании такого полезного материала.
Автор предлагает практические рекомендации, основанные на исследованиях и опыте.
Статья содержит разнообразные точки зрения, представленные в равной мере.
Автор статьи представляет разнообразные аспекты темы, предоставляя факты и аргументы без выражения собственного мнения.
Статья представляет несколько точек зрения на данную тему и анализирует их достоинства и недостатки. Это помогает читателю рассмотреть проблему с разных сторон и принять информированное решение.
Статья содержит практические рекомендации, которые можно применить в реальной жизни для решения проблемы.
This is very interesting, You are a very skilled blogger. I have joined your feed and look forward to seeking more of your magnificent post. Also, I’ve shared your website in my social networks!
What’s up to every , as I am truly keen of reading this website’s post to be updated daily. It contains fastidious material.
Я хотел бы выразить свою восторженность этой статьей! Она не только информативна, но и вдохновляет меня на дальнейшее изучение темы. Автор сумел передать свою страсть и знания, что делает эту статью поистине уникальной.
Автор предлагает аргументы, подкрепленные проверенными фактами и авторитетными источниками.
Эта статья является настоящим сокровищем информации. Я был приятно удивлен ее глубиной и разнообразием подходов к рассматриваемой теме. Спасибо автору за такой тщательный анализ и интересные факты!
Автор представляет разные точки зрения на проблему и оставляет читателю пространство для собственных размышлений.
Я хотел бы подчеркнуть четкость и последовательность изложения в этой статье. Автор сумел объединить информацию в понятный и логичный рассказ, что помогло мне лучше усвоить материал. Очень ценная статья!
What’s Going down i am new to this, I stumbled upon this I’ve found It positively helpful and it has aided me out loads. I hope to give a contribution & help other users like its helped me. Great job.
Я хотел бы отметить глубину исследования, представленную в этой статье. Автор не только предоставил факты, но и провел анализ их влияния и последствий. Это действительно ценный и информативный материал!
Inspiring story there. What happened after? Take care!
Magnificent goods from you, man. I have understand your stuff previous to and you’re just too wonderful. I actually like what you have acquired here, certainly like what you are saying and the way in which you say it. You make it entertaining and you still take care of to keep it smart. I cant wait to read much more from you. This is actually a terrific site.
Статья обладает нейтральным тоном и представляет различные точки зрения. Хорошо, что автор уделил внимание как плюсам, так и минусам рассматриваемой темы.
Автор статьи предоставляет информацию в понятной форме, избегая субъективных оценок.
I am curious to find out what blog platform you have been working with? I’m having some small security problems with my latest website and I would like to find something more risk-free. Do you have any suggestions?
Автор предоставляет разнообразные источники для более глубокого изучения темы.
Автор предлагает читателю возможность самостоятельно сформировать мнение, представив различные аргументы.
This design is spectacular! You obviously know how to keep a reader entertained. Between your wit and your videos, I was almost moved to start my own blog (well, almost…HaHa!) Wonderful job. I really enjoyed what you had to say, and more than that, how you presented it. Too cool!
Автор подходит к этому вопросу с нейтральной позицией, предоставляя достаточно информации для обсуждения.
Статья представляет широкий обзор темы, учитывая различные аспекты проблемы.
Thank you for the good writeup. It in fact was a amusement account it. Look advanced to far added agreeable from you! By the way, how could we communicate?
Hi there colleagues, its great post regarding teachingand completely defined, keep it up all the time.
When some one searches for his essential thing, therefore he/she desires to be available that in detail, therefore that thing is maintained over here.
Эта статья просто великолепна! Она представляет информацию в полном объеме и включает в себя практические примеры и рекомендации. Я нашел ее очень полезной и вдохновляющей. Большое спасибо автору за такую выдающуюся работу!
Автор предлагает обоснованные и логические выводы на основе представленных фактов и данных.
Hi there, I found your site by way of Google even as searching for a similar topic, your website got here up, it looks good. I’ve bookmarked it in my google bookmarks.
Я хотел бы выразить признательность автору этой статьи за его объективный подход к теме. Он представил разные точки зрения и аргументы, что позволило мне получить полное представление о рассматриваемой проблеме. Очень впечатляюще!
If some one wants to be updated with latest technologies therefore he must be pay a quick visit this site and be up to date all the time.
Статья обладает нейтральным тоном и представляет различные точки зрения. Хорошо, что автор уделил внимание как плюсам, так и минусам рассматриваемой темы.
Автор статьи представляет информацию с акцентом на объективность и достоверность.
Я хотел бы поблагодарить автора этой статьи за его основательное исследование и глубокий анализ. Он представил информацию с обширной перспективой и помог мне увидеть рассматриваемую тему с новой стороны. Очень впечатляюще!
Автор старается быть нейтральным, чтобы читатели могли самостоятельно рассмотреть различные аспекты темы.
Статья предлагает обширный обзор темы, представляя разные точки зрения и аргументы.
Автор статьи представляет информацию в объективной манере, избегая субъективных оценок.
Мне понравилось разнообразие и глубина исследований, представленных в статье.
I just like the valuable information you provide on your articles. I’ll bookmark your weblog and test again here frequently. I’m rather certain I’ll learn a lot of new stuff proper right here! Best of luck for the following!
I constantly emailed this website post page to all my contacts, since if like to read it then my contacts will too.
It’s perfect time to make some plans for the future and it’s time to be happy. I have read this post and if I could I desire to suggest you some interesting things or advice. Maybe you could write next articles referring to this article. I desire to read even more things about it!
Мне понравилась глубина исследования, представленная в статье.
Автор предлагает рациональные и аргументированные выводы на основе представленных фактов.
Статья представляет широкий спектр точек зрения на проблему, что способствует более глубокому пониманию.
Информационная статья содержит полезный контент и представляет различные аспекты обсуждаемой темы.
Автор старается представить материал нейтрально, оставляя пространство для собственного рассмотрения и анализа.
Эта статья превзошла мои ожидания! Она содержит обширную информацию, иллюстрирует примерами и предлагает практические советы. Я благодарен автору за его усилия в создании такого полезного материала.
Автор предоставляет дополнительные ресурсы для тех, кто хочет углубиться в изучение темы.
Автор статьи представляет информацию без предвзятости, предоставляя различные точки зрения и факты.
Это помогает читателям получить полную картину и сделать собственные выводы.
Статья содержит достаточно информации для того, чтобы читатель мог сделать собственные выводы.
At this time it seems like Drupal is the top blogging platform out there right now. (from what I’ve read) Is that what you are using on your blog?
Hello there, You have performed an incredible job. I’ll definitely digg it and for my part suggest to my friends. I am confident they will be benefited from this website.
Я оцениваю использование автором разнообразных источников, что позволяет получить всестороннюю информацию.
Очень понятная и информативная статья! Автор сумел объяснить сложные понятия простым и доступным языком, что помогло мне лучше усвоить материал. Огромное спасибо за такое ясное изложение!
This paragraph provides clear idea in support of the new users of blogging, that actually how to do blogging.
This is a great tip particularly to those fresh to the blogosphere. Simple but very accurate info… Thank you for sharing this one. A must read post!
Статья содержит аналитический подход к проблеме и представляет разнообразные точки зрения.
Читателям предоставляется возможность обдумать и обсудить представленные факты и аргументы.
Автор старается быть балансированным, предоставляя достаточно контекста и фактов для полного понимания читателями.
You should be a part of a contest for one of the greatest websites on the net. I most certainly will recommend this web site!
Hello everybody, here every person is sharing such knowledge, therefore it’s good to read this web site, and I used to pay a visit this weblog everyday.
This is a very good tip especially to those fresh to the blogosphere. Brief but very precise info… Many thanks for sharing this one. A must read article!
Хорошо, что автор обратил внимание на различные аспекты данной проблемы.
Hi there, after reading this amazing post i am as well glad to share my know-how here with colleagues.
Greetings from Idaho! I’m bored to death at work so I decided to browse your blog on my iphone during lunch break. I enjoy the knowledge you provide here and can’t wait to take a look when I get home. I’m amazed at how fast your blog loaded on my cell phone .. I’m not even using WIFI, just 3G .. Anyways, very good site!
Я хотел бы отметить глубину исследования, представленную в этой статье. Автор не только предоставил факты, но и провел анализ их влияния и последствий. Это действительно ценный и информативный материал!
Я нашел в статье несколько полезных советов.
Эта статья – настоящий кладезь информации! Я оцениваю ее полноту и разнообразие представленных фактов. Автор сделал тщательное исследование и предоставил нам ценный ресурс для изучения темы. Большое спасибо за такое ценное содержание!
Автор статьи представляет информацию с акцентом на факты и статистику, не высказывая предпочтений.
We stumbled over here coming from a different page and thought I should check things out. I like what I see so now i’m following you. Look forward to looking over your web page repeatedly.
It’s an awesome paragraph designed for all the online viewers; they will get benefit from it I am sure.
My family members every time say that I am killing my time here at web, except I know I am getting experience all the time by reading thes nice content.
Статья содержит обширный объем информации, которая подкреплена соответствующими доказательствами.
Эта статья является настоящим источником вдохновения и мотивации. Она не только предоставляет информацию, но и стимулирует к дальнейшему изучению темы. Большое спасибо автору за его старания в создании такого мотивирующего контента!
Quality articles or reviews is the secret to interest the people to pay a visit the site, that’s what this web page is providing.
I enjoy reading a post that can make people think. Also, thanks for allowing me to comment!
You’ve made some good points there. I checked on the internet for more info about the issue and found most individuals will go along with your views on this site.
Автор предлагает анализ преимуществ и недостатков разных подходов к решению проблемы.
Автор предлагает аргументы, подкрепленные проверенными фактами и авторитетными источниками.
Отлично, что автор обратил внимание на разные точки зрения и представил их в сбалансированном виде.
It’s remarkable in favor of me to have a web site, which is helpful in favor of my knowledge. thanks admin
Я благодарен автору этой статьи за его тщательное и глубокое исследование. Он представил информацию с большой детализацией и аргументацией, что делает эту статью надежным источником знаний. Очень впечатляющая работа!
Читателям предоставляется возможность ознакомиться с различными аспектами темы и сделать собственные выводы.
Статья содержит систематическую аналитику темы, учитывая разные аспекты проблемы.
Автор старается предоставить достоверную информацию, не влияя на оценку читателей. Это сообщение отправлено с сайта https://ru.gototop.ee/
Автор предлагает практические советы, которые читатели могут использовать в своей повседневной жизни.
Very good write-up. I certainly appreciate this website. Continue the good work!
Hello there! I could have sworn I’ve been to this site before but after going through a few of the posts I realized it’s new to me. Regardless, I’m certainly pleased I stumbled upon it and I’ll be bookmarking it and checking back often!
Я оцениваю тщательность и точность исследования, представленного в этой статье. Автор провел глубокий анализ и представил аргументированные выводы. Очень важная и полезная работа!
We are a group of volunteers and starting a new scheme in our community. Your site offered us with valuable info to work on. You’ve done an impressive job and our entire community will be grateful to you.
These are genuinely wonderful ideas in regarding blogging. You have touched some nice points here. Any way keep up wrinting.
Great post. I’m experiencing a few of these issues as well..
Я оцениваю умение автора объединить разные точки зрения и синтезировать их в понятную картину.
Статья представляет разные стороны дискуссии, не выражая предпочтений или приоритетов.
Thanks for the good writeup. It if truth be told was a leisure account it. Glance complicated to far brought agreeable from you! However, how could we communicate?
Appreciation to my father who shared with me regarding this blog, this weblog is in fact awesome.
Мне понравилась систематическая структура статьи, которая позволяет читателю легко следовать логике изложения.
Good response in return of this matter with firm arguments and telling the whole thing about that.
Мне понравилась объективность и сбалансированность в подаче материала в статье.
Статья содержит ссылки на актуальные и авторитетные источники, что делает ее надежной и достоверной.
When I initially commented I clicked the “Notify me when new comments are added” checkbox and now each time a comment is added I get three e-mails with the same comment. Is there any way you can remove people from that service? Many thanks!
Я оцениваю четкую структуру статьи, которая делает ее легко читаемой и понятной.
Автор старается быть нейтральным, предоставляя читателям возможность самих оценить представленные доводы.
Автор предоставляет примеры и иллюстрации, чтобы проиллюстрировать свои аргументы и упростить понимание темы.
Автор статьи предоставляет различные точки зрения и экспертные мнения, не принимая сторону.
Very good post. I definitely appreciate this website. Keep it up!
This design is spectacular! You obviously know how to keep a reader amused. Between your wit and your videos, I was almost moved to start my own blog (well, almost…HaHa!) Fantastic job. I really loved what you had to say, and more than that, how you presented it. Too cool!
Heya i am for the first time here. I found this board and I find It really useful & it helped me out a lot. I hope to give something back and help others like you aided me.
Hello There. I found your blog using msn. This is an extremely well written article. I will make sure to bookmark it and return to read more of your useful info. Thanks for the post. I will definitely comeback.
Hi there, this weekend is pleasant in favor of me, because this time i am reading this fantastic informative article here at my house.
This article will assist the internet viewers for setting up new weblog or even a weblog from start to end.
Я оцениваю использование автором разнообразных источников, что позволяет получить всестороннюю информацию.
Автор предлагает читателю дополнительные ресурсы для более глубокого изучения темы.
Я хотел бы выразить признательность автору этой статьи за его объективный подход к теме. Он представил разные точки зрения и аргументы, что позволило мне получить полное представление о рассматриваемой проблеме. Очень впечатляюще!
Очень хорошо исследованная статья! Она содержит много подробностей и является надежным источником информации. Я оцениваю автора за его тщательную работу и приветствую его старания в предоставлении читателям качественного контента.
Я чувствую, что эта статья является настоящим источником вдохновения. Она предлагает новые идеи и вызывает желание узнать больше. Большое спасибо автору за его творческий и информативный подход!
Статья содержит анализ причин и последствий проблемы, что позволяет лучше понять ее важность и сложность.
Автор статьи предоставляет информацию, подкрепленную надежными источниками, что делает ее достоверной и нейтральной.
Amazing! This blog looks just like my old one! It’s on a completely different topic but it has pretty much the same layout and design. Great choice of colors!
It’s an remarkable post designed for all the internet people; they will obtain benefit from it I am sure.
Статья содержит аргументы, подкрепленные реальными примерами и исследованиями.
Статья предлагает объективный обзор темы, предоставляя аргументы и контекст.
Отличная статья! Я бы хотел отметить ясность и логичность, с которыми автор представил информацию. Это помогло мне легко понять сложные концепции. Большое спасибо за столь прекрасную работу!
Я прочитал эту статью с большим удовольствием! Она написана ясно и доступно, несмотря на сложность темы. Большое спасибо автору за то, что делает сложные понятия понятными для всех.
Good day! Would you mind if I share your blog with my facebook group? There’s a lot of people that I think would really appreciate your content. Please let me know. Cheers
Статья содержит обширную информацию и аргументы, подтвержденные ссылками на достоверные источники.
Автор предоставляет подробные сведения и контекст, что помогает читателям лучше понять обсуждаемую тему.
When I originally commented I clicked the “Notify me when new comments are added” checkbox and now each time a comment is added I get several e-mails with the same comment. Is there any way you can remove me from that service? Cheers!
Статья представляет несколько точек зрения на данную тему и анализирует их достоинства и недостатки. Это помогает читателю рассмотреть проблему с разных сторон и принять информированное решение.
Надеюсь, что эти дополнительные комментарии принесут ещё больше позитивных отзывов на информационную статью!
Я благодарен автору этой статьи за его тщательное и глубокое исследование. Он представил информацию с большой детализацией и аргументацией, что делает эту статью надежным источником знаний. Очень впечатляющая работа!
Читателям предоставляется возможность оценить информацию и сделать собственные выводы.
WOW just what I was looking for. Came here by searching for keyword
Очень понятная и информативная статья! Автор сумел объяснить сложные понятия простым и доступным языком, что помогло мне лучше усвоить материал. Огромное спасибо за такое ясное изложение!
Автор предлагает анализ различных точек зрения на проблему без призыва к одной конкретной позиции.
Эта статья является примером качественного исследования и профессионализма. Автор предоставил нам широкий обзор темы и представил информацию с точки зрения эксперта. Очень важный вклад в популяризацию знаний!
Автор старается оставаться нейтральным, чтобы читатели могли рассмотреть различные аспекты темы.
Я хотел бы подчеркнуть четкость и последовательность изложения в этой статье. Автор сумел объединить информацию в понятный и логичный рассказ, что помогло мне лучше усвоить материал. Очень ценная статья!
These are genuinely wonderful ideas in concerning blogging. You have touched some good factors here. Any way keep up wrinting.
Статья содержит систематическую аналитику темы, учитывая разные аспекты проблемы.
Я хотел бы выразить свою восторженность этой статьей! Она не только информативна, но и вдохновляет меня на дальнейшее изучение темы. Автор сумел передать свою страсть и знания, что делает эту статью поистине уникальной.
Это позволяет читателям самостоятельно сформировать свое мнение и оценить представленные доводы.
Информационная статья содержит полезный контент и представляет различные аспекты обсуждаемой темы.
Мне понравилась четкость и логика аргументации автора в статье.
Автор предлагает анализ преимуществ и недостатков различных решений, связанных с темой.
I don’t even understand how I finished up right here, but I thought this publish used to be good. I don’t realize who you’re but definitely you’re going to a well-known blogger in the event you aren’t already. Cheers!
These are truly wonderful ideas in on the topic of blogging. You have touched some nice points here. Any way keep up wrinting.
Автор статьи предоставляет разнообразные источники и мнения экспертов, не принимая определенную позицию.
Hi, i read your blog occasionally and i own a similar one and i was just wondering if you get a lot of spam feedback? If so how do you reduce it, any plugin or anything you can recommend? I get so much lately it’s driving me mad so any support is very much appreciated.
Автор приводит примеры из различных источников, что позволяет получить более полное представление о теме. Статья является нейтральным и информативным ресурсом для тех, кто интересуется данной проблематикой.
Автор не высказывает собственных предпочтений, что позволяет читателям самостоятельно сформировать свое мнение.
I’m really impressed together with your writing abilities as smartly as with the structure for your blog. Is this a paid theme or did you modify it yourself? Either way stay up the excellent quality writing, it’s rare to peer a nice weblog like this one these days..
Я хотел бы выразить свою восторженность этой статьей! Она не только информативна, но и вдохновляет меня на дальнейшее изучение темы. Автор сумел передать свою страсть и знания, что делает эту статью поистине уникальной.
Очень интересная исследовательская работа! Статья содержит актуальные факты, аргументированные доказательствами. Это отличный источник информации для всех, кто хочет поглубже изучить данную тему.
Я оцениваю объективность и сбалансированность подхода автора к представлению информации.
You really make it appear so easy together with your presentation but I find this matter to be actually something which I feel I’d never understand. It seems too complicated and very extensive for me. I’m looking forward in your subsequent publish, I’ll attempt to get the hang of it!
Автор предлагает анализ преимуществ и недостатков разных подходов к решению проблемы.
Автор предлагает объективный анализ различных решений, связанных с проблемой.
This design is wicked! You certainly know how to keep a reader amused. Between your wit and your videos, I was almost moved to start my own blog (well, almost…HaHa!) Fantastic job. I really loved what you had to say, and more than that, how you presented it. Too cool!
Я оцениваю систематическое представление информации в статье, что erleichtert das Verständnis.
My developer is trying to convince me to move to .net from PHP. I have always disliked the idea because of the expenses. But he’s tryiong none the less. I’ve been using WordPress on a variety of websites for about a year and am concerned about switching to another platform. I have heard excellent things about blogengine.net. Is there a way I can import all my wordpress posts into it? Any kind of help would be really appreciated!
Позиция автора не является однозначной, что позволяет читателям более глубоко разобраться в обсуждаемой теме.
Статья представляет все основные аспекты темы, без излишней детализации.
Я хотел бы выразить свою благодарность автору за его глубокие исследования и ясное изложение. Он сумел объединить сложные концепции и представить их в доступной форме. Это действительно ценный ресурс для всех, кто интересуется этой темой.
Автор предлагает анализ плюсов и минусов разных подходов к решению проблемы.
Howdy! This is kind of off topic but I need some advice from an established blog. Is it difficult to set up your own blog? I’m not very techincal but I can figure things out pretty fast. I’m thinking about creating my own but I’m not sure where to start. Do you have any points or suggestions? Cheers
Статья предлагает широкий обзор событий и фактов, связанных с обсуждаемой темой.
Спасибо за эту статью! Она превзошла мои ожидания. Информация была представлена кратко и ясно, и я оставил эту статью с более глубоким пониманием темы. Отличная работа!
I have to thank you for the efforts you have put in writing this site. I am hoping to check out the same high-grade content from you later on as well. In truth, your creative writing abilities has motivated me to get my own, personal website now 😉
Я бы хотел выразить свою благодарность автору этой статьи за его профессионализм и преданность точности. Он предоставил достоверные факты и аргументированные выводы, что делает эту статью надежным источником информации.
Ridiculous story there. What happened after? Good luck!
Статья предлагает обширный обзор темы, представляя разные точки зрения и аргументы.
I’m truly enjoying the design and layout of your blog. It’s a very easy on the eyes which makes it much more enjoyable for me to come here and visit more often. Did you hire out a developer to create your theme? Exceptional work!
What’s up to all, how is everything, I think every one is getting more from this website, and your views are nice in favor of new users.
Это помогает создать обстановку для объективного обсуждения.
Автор статьи предоставляет информацию, основанную на различных источниках и экспертных мнениях.
Does your website have a contact page? I’m having problems locating it but, I’d like to send you an e-mail. I’ve got some ideas for your blog you might be interested in hearing. Either way, great blog and I look forward to seeing it develop over time.
Автор предлагает логические выводы на основе представленных фактов и аргументов.
Статья предлагает глубокий анализ темы и рассматривает ее со всех сторон.
Информационная статья предлагает взвешенный подход к обсуждаемой теме, аргументируя свои выводы доказательствами и статистикой.
Автор статьи представляет разнообразные аспекты темы, предоставляя факты и аргументы без выражения собственного мнения.
Hello! Quick question that’s entirely off topic. Do you know how to make your site mobile friendly? My weblog looks weird when browsing from my iphone 4. I’m trying to find a theme or plugin that might be able to correct this issue. If you have any suggestions, please share. With thanks!
Увеличение ссылочной массы: Ключевой фактор в росте DR сайта. Увеличение ссылочной массы является неотъемлемой частью стратегии роста DR сайта. Этот процесс требует систематического и длительного подхода, включающего в себя поиск высококачественных ссылок, мониторинг и анализ ссылочной массы, а также адаптацию к постоянно меняющемуся окружению поисковых систем. Понимание этих аспектов и их правильная реализация помогут сайту достичь более высокого DR, что в свою очередь улучшит его ранжирование, привлечет больше органического трафика и повысит успех онлайн-присутствия бизнеса.
Я восхищен тем, как автор умело объясняет сложные концепции. Он сумел сделать информацию доступной и интересной для широкой аудитории. Это действительно заслуживает похвалы!
Hi, its nice piece of writing on the topic of media print, we all know media is a fantastic source of information.
Это позволяет читателям самостоятельно сформировать свое мнение.
Hi there to every single one, it’s genuinely a good for me to pay a visit this site, it includes useful Information.
Статья помогла мне лучше понять сложные концепции, связанные с темой.
I do not know if it’s just me or if perhaps everyone else experiencing problems with your blog. It appears as if some of the written text in your posts are running off the screen. Can someone else please provide feedback and let me know if this is happening to them too? This might be a issue with my internet browser because I’ve had this happen previously. Many thanks
great post, very informative. I wonder why the other specialists of this sector don’t understand this. You must proceed your writing. I’m sure, you’ve a great readers’ base already!
Because the admin of this web page is working, no hesitation very soon it will be famous, due to its feature contents.
Автор старается представить информацию нейтрально и всеобъемлюще.
I got this web site from my buddy who shared with me on the topic of this website and at the moment this time I am visiting this web page and reading very informative articles at this place.
This website certainly has all of the information I needed concerning this subject and didn’t know who to ask.
Статья предлагает разнообразные данные и факты, представленные без пристрастия.
Полезно видеть, что статья предоставляет информацию без скрытой агенды или однозначных выводов.
Надеюсь, что эти дополнительные комментарии принесут ещё больше позитивных отзывов на информационную статью!
Статья содержит обоснованные аргументы, подкрепленные фактами.
Мне понравилась объективность автора и его стремление представить все стороны вопроса.
This article provides clear idea in support of the new visitors of blogging, that genuinely how to do blogging.
I like the valuable info you provide in your articles. I will bookmark your blog and check again here frequently. I am quite sure I’ll learn lots of new stuff right here! Best of luck for the next!
Howdy just wanted to give you a quick heads up and let you know a few of the pictures aren’t loading properly. I’m not sure why but I think its a linking issue. I’ve tried it in two different browsers and both show the same outcome.
Мне понравилась аргументация автора, основанная на логической цепочке рассуждений.
Эта статья – источник вдохновения и новых знаний! Я оцениваю уникальный подход автора и его способность представить информацию в увлекательной форме. Это действительно захватывающее чтение!
Greetings! I know this is somewhat off topic but I was wondering which blog platform are you using for this website? I’m getting sick and tired of WordPress because I’ve had problems with hackers and I’m looking at options for another platform. I would be great if you could point me in the direction of a good platform.
Автор предлагает анализ преимуществ и недостатков разных подходов к решению проблемы.
Эта статья является настоящим источником вдохновения и мотивации. Она не только предоставляет информацию, но и стимулирует к дальнейшему изучению темы. Большое спасибо автору за его старания в создании такого мотивирующего контента!
Я восхищен тем, как автор умело объясняет сложные концепции. Он сумел сделать информацию доступной и интересной для широкой аудитории. Это действительно заслуживает похвалы!
Статья содержит информацию, основанную на достоверных источниках и экспертных мнениях.
Hi there! I know this is somewhat off topic but I was wondering which blog platform are you using for this site? I’m getting sick and tired of WordPress because I’ve had problems with hackers and I’m looking at options for another platform. I would be great if you could point me in the direction of a good platform.
Я оцениваю широту покрытия темы в статье.
excellent points altogether, you simply won a emblem new reader. What might you suggest about your post that you simply made some days ago? Any positive?
Это помогает читателям получить полную картину и сделать собственные выводы.
Very good information. Lucky me I ran across your site by accident (stumbleupon). I have book-marked it for later!
Автор старается быть балансированным, предоставляя достаточно контекста и фактов для полного понимания читателями.
Эта статья – источник вдохновения и новых знаний! Я оцениваю уникальный подход автора и его способность представить информацию в увлекательной форме. Это действительно захватывающее чтение!
Автор предлагает практические советы, которые читатели могут использовать в своей повседневной жизни.
Это помогает читателям получить полное представление о спорной проблеме.
Hello! Do you use Twitter? I’d like to follow you if that would be ok. I’m definitely enjoying your blog and look forward to new updates.
Hi to every one, for the reason that I am truly eager of reading this web site’s post to be updated regularly. It contains fastidious information.
What’s up, for all time i used to check blog posts here in the early hours in the morning, since i enjoy to gain knowledge of more and more.
Полезная информация для тех, кто стремится получить всестороннее представление.
Я прочитал эту статью с огромным интересом! Автор умело объединил факты, статистику и персональные истории, что делает ее настоящей находкой. Я получил много новых знаний и вдохновения. Браво!
Я чувствую, что эта статья является настоящим источником вдохновения. Она предлагает новые идеи и вызывает желание узнать больше. Большое спасибо автору за его творческий и информативный подход!
Hello! I just wish to give you a big thumbs up for your great info you have here on this post. I’ll be coming back to your website for more soon.
Эта статья является примером качественного исследования и профессионализма. Автор предоставил нам широкий обзор темы и представил информацию с точки зрения эксперта. Очень важный вклад в популяризацию знаний!
Автор старается подойти к теме без предубеждений.
Just desire to say your article is as amazing. The clearness in your post is just nice and i could assume you are an expert on this subject. Fine with your permission allow me to grab your RSS feed to keep updated with forthcoming post. Thanks a million and please keep up the gratifying work.
continuously i used to read smaller content that as well clear their motive, and that is also happening with this article which I am reading here.
Надеюсь, что эти дополнительные комментарии принесут ещё больше позитивных отзывов на информационную статью!
Very good info. Lucky me I discovered your blog by accident (stumbleupon). I have saved as a favorite for later!
My brother recommended I might like this web site. He was once entirely right. This post actually made my day. You cann’t imagine just how much time I had spent for this information! Thank you!
Статья представляет все основные аспекты темы, без излишней детализации.
I am not certain the place you are getting your info, but great topic. I must spend a while studying more or figuring out more. Thanks for fantastic information I was in search of this info for my mission.
What a material of un-ambiguity and preserveness of valuable experience concerning unpredicted feelings.
It’s remarkable in support of me to have a web page, which is helpful in favor of my know-how. thanks admin
Thanks in support of sharing such a good idea, post is nice, thats why i have read it entirely
An impressive share! I have just forwarded this onto a colleague who had been conducting a little research on this. And he in fact bought me dinner simply because I found it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanks for spending the time to talk about this subject here on your blog.
What’s up, yup this paragraph is in fact nice and I have learned lot of things from it on the topic of blogging. thanks.
Автор статьи умело анализирует сложные концепции и представляет их в понятной форме.
Simply wish to say your article is as astonishing. The clarity to your put up is just excellent and that i could assume you are knowledgeable on this subject. Well together with your permission let me to take hold of your RSS feed to keep up to date with imminent post. Thanks one million and please continue the enjoyable work.
Thank you for the good writeup. It in fact was a amusement account it. Look advanced to far added agreeable from you! However, how can we communicate?
I visited multiple web pages however the audio quality for audio songs current at this web page is really wonderful.
Автор предоставляет подробные сведения и контекст, что помогает читателям лучше понять обсуждаемую тему.
When I initially commented I clicked the “Notify me when new comments are added” checkbox and now each time a comment is added I get several emails with the same comment. Is there any way you can remove people from that service? Many thanks!
Я оцениваю объективный подход автора и его стремление представить полную картину проблемы.
Автор статьи представляет информацию без предвзятости, предоставляя различные точки зрения и факты.
Have you ever thought about publishing an e-book or guest authoring on other websites? I have a blog centered on the same topics you discuss and would love to have you share some stories/information. I know my viewers would enjoy your work. If you’re even remotely interested, feel free to send me an e mail.
With havin so much content and articles do you ever run into any issues of plagorism or copyright violation? My site has a lot of exclusive content I’ve either created myself or outsourced but it seems a lot of it is popping it up all over the internet without my agreement. Do you know any methods to help prevent content from being ripped off? I’d genuinely appreciate it.
Автор предоставляет ссылки на авторитетные источники, что делает статью надежной и достоверной.
Статья содержит актуальную информацию, которая помогает понять сложность и важность проблемы.
you’re actually a just right webmaster. The website loading velocity is amazing. It seems that you are doing any unique trick. In addition, The contents are masterpiece. you’ve performed a excellent job on this subject!
Эта статья является примером качественного исследования и профессионализма. Автор предоставил нам широкий обзор темы и представил информацию с точки зрения эксперта. Очень важный вклад в популяризацию знаний!
Эта статья – настоящий кладезь информации! Я оцениваю ее полноту и разнообразие представленных фактов. Автор сделал тщательное исследование и предоставил нам ценный ресурс для изучения темы. Большое спасибо за такое ценное содержание!
Hmm it appears like your blog ate my first comment (it was extremely long) so I guess I’ll just sum it up what I wrote and say, I’m thoroughly enjoying your blog. I as well am an aspiring blog writer but I’m still new to everything. Do you have any tips and hints for rookie blog writers? I’d certainly appreciate it.
Автор использовал разнообразные источники, чтобы подкрепить свои утверждения.
Эта статья является настоящим сокровищем информации. Я был приятно удивлен ее глубиной и разнообразием подходов к рассматриваемой теме. Спасибо автору за такой тщательный анализ и интересные факты!
Wow, that’s what I was exploring for, what a material! present here at this blog, thanks admin of this web page.
I know this if off topic but I’m looking into starting my own weblog and was curious what all is needed to get setup? I’m assuming having a blog like yours would cost a pretty penny? I’m not very internet smart so I’m not 100 positive. Any suggestions or advice would be greatly appreciated. Thank you
Это помогает создать обстановку, в которой читатели могут обдумать и обсудить тему без пристрастия.
Informative article, exactly what I was looking for.
Автор представляет информацию в организованной и последовательной форме, что erleichtert das Verständnis.
Я восхищен тем, как автор умело объясняет сложные концепции. Он сумел сделать информацию доступной и интересной для широкой аудитории. Это действительно заслуживает похвалы!
Статья предоставляет информацию из разных источников, обеспечивая балансированное представление фактов и аргументов.
Автор старается подойти к теме без предубеждений.
Hello there, You have performed an incredible job. I will certainly digg it and personally suggest to my friends. I’m confident they will be benefited from this web site.
Статья предлагает обширный обзор темы, представляя разные точки зрения и аргументы.
Great work! This is the type of information that should be shared across the web. Disgrace on the seek engines for now not positioning this publish upper! Come on over and seek advice from my web site . Thank you =)
Автор старается сохранить нейтральность, предоставляя обстоятельную основу для дальнейшего рассмотрения темы.
Читателям предоставляется возможность самостоятельно изучить представленные факты и сделать информированный вывод.
Hey there! Do you know if they make any plugins to protect against hackers? I’m kinda paranoid about losing everything I’ve worked hard on. Any suggestions?
Я очень доволен, что прочитал эту статью. Она оказалась настоящим открытием для меня. Информация была представлена в увлекательной и понятной форме, и я получил много новых знаний. Спасибо автору за такое удивительное чтение!
Автор старается быть нейтральным, что помогает читателям лучше понять обсуждаемую тему.
When I originally commented I clicked the “Notify me when new comments are added” checkbox and now each time a comment is added I get four emails with the same comment. Is there any way you can remove me from that service? Many thanks!
Это помогает читателям получить всестороннее представление о теме без явных предубеждений.
Статья содержит разнообразные факты и аргументы, представленные в объективной манере.
Автор предоставляет разнообразные источники, которые дополняют и расширяют представленную информацию.
Статья представляет разнообразные аргументы и позиции, основанные на существующих данных и экспертном мнении.
Мне понравилась объективность и сбалансированность в подаче материала в статье.
Конечно, вот ещё несколько положительных комментариев на информационную статью: Это сообщение отправлено с сайта GoToTop.ee
Я оцениваю объективность и сбалансированность аргументации в статье.
Автор предоставляет достаточно информации, чтобы читатель мог составить собственное мнение по данной теме.
With havin so much content and articles do you ever run into any problems of plagorism or copyright infringement? My site has a lot of completely unique content I’ve either created myself or outsourced but it looks like a lot of it is popping it up all over the internet without my permission. Do you know any techniques to help protect against content from being ripped off? I’d really appreciate it.
Статья содержит обоснованные аргументы, подкрепленные фактами.
Читателям предоставляется возможность ознакомиться с разными точками зрения и самостоятельно сформировать свое мнение.
Автор приводит конкретные примеры, чтобы проиллюстрировать свои аргументы.
This website was… how do I say it? Relevant!! Finally I’ve found something which helped me. Cheers!
Статья предоставляет объективную информацию о теме, подкрепленную различными источниками.
Hi everyone, it’s my first go to see at this web page, and post is in fact fruitful designed for me, keep up posting these types of content.
Это помогает читателям осознать сложность проблемы и самостоятельно сформировать свое собственное мнение.
Это позволяет читателям самостоятельно сформировать свое мнение.
Аргументы подкреплены фактами и исследованиями, что позволяет читателям рассмотреть разные стороны вопроса.
Why people still use to read news papers when in this technological globe the whole thing is accessible on net?
Информационная статья представляет различные аргументы и контекст в отношении обсуждаемой темы.
Автор предлагает практические рекомендации, основанные на исследованиях и опыте.
Hi, I do believe this is an excellent blog. I stumbledupon it 😉 I am going to revisit yet again since i have book marked it. Money and freedom is the greatest way to change, may you be rich and continue to guide other people.
Мне понравилась систематическая структура статьи, которая позволяет читателю легко следовать логике изложения.
Your way of describing everything in this paragraph is in fact pleasant, every one can easily be aware of it, Thanks a lot.
Hey there, You’ve done an incredible job. I’ll definitely digg it and individually suggest to my friends. I’m confident they’ll be benefited from this web site.
Я хотел бы отметить глубину исследования, представленную в этой статье. Автор не только предоставил факты, но и провел анализ их влияния и последствий. Это действительно ценный и информативный материал!
Статья содержит сбалансированный подход к теме и учитывает различные точки зрения.
Hello friends, its wonderful article on the topic of cultureand fully defined, keep it up all the time.
Статья содержит актуальную статистику, что помогает оценить масштаб проблемы.
Статья является информативной и предоставляет различные факты и аргументы.
Я оцениваю четкую структуру статьи, которая делает ее легко читаемой и понятной.
Thank you a lot for sharing this with all people you actually recognize what you are talking approximately! Bookmarked. Please also consult with my website =). We can have a link change contract among us
Simply wish to say your article is as astounding. The clearness on your put up is just spectacular and that i could suppose you’re a professional in this subject. Fine with your permission let me to grasp your RSS feed to stay up to date with impending post. Thanks 1,000,000 and please keep up the gratifying work.
Мне понравилась глубина исследования, представленная в статье.
Wonderful article! That is the kind of info that are supposed to be shared across the web. Disgrace on the seek engines for now not positioning this post upper! Come on over and talk over with my website . Thanks =)
Статья предлагает разнообразные подходы к решению проблемы и позволяет читателю выбрать наиболее подходящий для него.
Статья содержит полезную информацию, которая может быть полезной для практического применения.
Автор статьи представляет информацию с акцентом на объективность и достоверность.
Unquestionably believe that which you said. Your favorite justification seemed to be on the internet the easiest thing to be aware of. I say to you, I definitely get irked while people consider worries that they just do not know about. You managed to hit the nail upon the top and also defined out the whole thing without having side-effects , people could take a signal. Will probably be back to get more. Thanks
Мне понравилась четкая и последовательная логика аргументации автора в статье.
Статья содержит аргументы, которые помогают читателю лучше понять важность и последствия проблемы.
Hi there, just changed into alert to your blog through Google, and located that it is truly informative. I’m gonna watch out for brussels. I will appreciate if you happen to continue this in future. Lots of folks shall be benefited from your writing. Cheers!
Я не могу не отметить качество исследования, представленного в этой статье. Автор использовал надежные источники и предоставил нам актуальную информацию. Большое спасибо за такой надежный и информативный материал!
Статья содержит сбалансированный подход к теме и учитывает различные точки зрения.
Hi, I wish for to subscribe for this blog to take hottest updates, so where can i do it please assist.
Статья содержит аргументы, которые помогают читателю лучше понять важность и последствия проблемы.
Я рад, что наткнулся на эту статью. Она содержит уникальные идеи и интересные точки зрения, которые позволяют глубже понять рассматриваемую тему. Очень познавательно и вдохновляюще!
Автор предлагает подробное объяснение сложных понятий, связанных с темой.
Статья предлагает обширный обзор темы, представляя разные точки зрения и аргументы.
Автор статьи предоставляет информацию в понятной форме, избегая субъективных оценок.
My family members every time say that I am killing my time here at web, except I know I am getting experience daily by reading thes fastidious articles.
Статья предлагает читателю возможность самостоятельно сформировать свое мнение на основе представленных аргументов.
Marvelous, what a weblog it is! This website provides helpful information to us, keep it up.
Yesterday, while I was at work, my sister stole my iPad and tested to see if it can survive a 30 foot drop, just so she can be a youtube sensation. My apple ipad is now broken and she has 83 views. I know this is totally off topic but I had to share it with someone!
I every time emailed this website post page to all my friends, because if like to read it next my contacts will too.
I quite like reading through an article that can make people think. Also, thank you for permitting me to comment!
This is a topic which is close to my heart… Take care! Exactly where are your contact details though?
Oh my goodness! Amazing article dude! Thanks, However I am going through difficulties with your RSS. I don’t know the reason why I am unable to subscribe to it. Is there anyone else getting the same RSS issues? Anybody who knows the solution will you kindly respond? Thanks!!
I don’t even know how I ended up here, but I thought this post was great. I don’t know who you are but definitely you are going to a famous blogger if you are not already 😉 Cheers!
Статья содержит ссылки на актуальные и авторитетные источники, что делает ее надежной и достоверной.
Статья основана на объективных данных и исследованиях.
This is a topic that’s near to my heart… Cheers! Exactly where are your contact details though?
Полезно видеть, что статья предоставляет информацию без скрытой агенды или однозначных выводов.
Hi there, always i used to check website posts here in the early hours in the dawn, because i enjoy to find out more and more.
Эта статья действительно заслуживает высоких похвал! Она содержит информацию, которую я долго искал, и дает полное представление о рассматриваемой теме. Благодарю автора за его тщательную работу и отличное качество материала!
Это позволяет читателям получить разностороннюю информацию и самостоятельно сделать выводы.
Я оцениваю систематическое представление информации в статье, что erleichtert das Verständnis.
Я оцениваю широту охвата темы в статье.
Эта статья является настоящим источником вдохновения и мотивации. Она не только предоставляет информацию, но и стимулирует к дальнейшему изучению темы. Большое спасибо автору за его старания в создании такого мотивирующего контента!
Автор статьи предлагает достоверные данные и факты, представленные в нейтральном ключе.
Woah! I’m really enjoying the template/theme of this blog. It’s simple, yet effective. A lot of times it’s difficult to get that “perfect balance” between superb usability and visual appeal. I must say you have done a superb job with this. In addition, the blog loads extremely fast for me on Firefox. Exceptional Blog!
Я оцениваю объективность и сбалансированность аргументации в статье.
Heya this is kinda of off topic but I was wondering if blogs use WYSIWYG editors or if you have to manually code with HTML. I’m starting a blog soon but have no coding knowledge so I wanted to get guidance from someone with experience. Any help would be enormously appreciated!
Wow! After all I got a blog from where I be capable of in fact get useful data concerning my study and knowledge.
I am no longer sure the place you are getting your information, but great topic. I must spend a while studying more or understanding more. Thank you for magnificent information I was in search of this information for my mission.
Автор старается оставаться объективным, чтобы читатели могли оценить различные аспекты и сформировать собственное понимание. Это сообщение отправлено с сайта https://ru.gototop.ee/
Читателям предоставляется возможность самостоятельно сформировать свое мнение на основе представленных фактов.
Я оцениваю умение автора использовать разнообразные источники, чтобы подкрепить свои утверждения.
Очень интересная исследовательская работа! Статья содержит актуальные факты, аргументированные доказательствами. Это отличный источник информации для всех, кто хочет поглубже изучить данную тему.
I am not very good with English but I get hold this really easygoing to interpret.
Статья содержит ссылки на актуальные и авторитетные источники, что делает ее надежной и достоверной.
My spouse and I absolutely love your blog and find a lot of your post’s to be just what I’m looking for. Would you offer guest writers to write content available for you? I wouldn’t mind composing a post or elaborating on most of the subjects you write about here. Again, awesome site!
Эта статья – источник ценной информации! Я оцениваю глубину исследования и разнообразие рассматриваемых аспектов. Она действительно расширила мои знания и помогла мне лучше понять тему. Большое спасибо автору за такую качественную работу!
Great delivery. Outstanding arguments. Keep up the great spirit.
Quality content is the main to be a focus for the visitors to visit the web page, that’s what this web page is providing.
Have you ever thought about publishing an e-book or guest authoring on other websites? I have a blog based upon on the same subjects you discuss and would love to have you share some stories/information. I know my visitors would appreciate your work. If you are even remotely interested, feel free to send me an email.
Я ценю фактический и информативный характер этой статьи. Она предлагает читателю возможность рассмотреть различные аспекты рассматриваемой проблемы без внушения какого-либо определенного мнения.
It’s enormous that you are getting ideas from this piece of writing as well as from our argument made here.
Автор статьи предлагает разные точки зрения на обсуждаемую проблему, позволяя читателям ознакомиться с различными аргументами и сделать собственные выводы.
If you want to get a great deal from this post then you have to apply these strategies to your won web site.
Я хотел бы выразить свою восторженность этой статьей! Она не только информативна, но и вдохновляет меня на дальнейшее изучение темы. Автор сумел передать свою страсть и знания, что делает эту статью поистине уникальной.
Мне понравилась систематическая структура статьи, которая позволяет читателю легко следовать логике изложения.
Автор предлагает несколько точек зрения на проблему, что позволяет читателю сформировать свое мнение.
Tremendous things here. I’m very glad to peer your post. Thank you so much and I am having a look ahead to contact you. Will you please drop me a e-mail?
Hi there to all, it’s really a good for me to go to see this web page, it includes helpful Information.
Way cool! Some very valid points! I appreciate you writing this write-up and the rest of the site is really good.
Очень понятная и информативная статья! Автор сумел объяснить сложные понятия простым и доступным языком, что помогло мне лучше усвоить материал. Огромное спасибо за такое ясное изложение! Это сообщение отправлено с сайта https://ru.gototop.ee/
I’m really enjoying the design and layout of your blog. It’s a very easy on the eyes which makes it much more pleasant for me to come here and visit more often. Did you hire out a designer to create your theme? Outstanding work!
Hey I know this is off topic but I was wondering if you knew of any widgets I could add to my blog that automatically tweet my newest twitter updates. I’ve been looking for a plug-in like this for quite some time and was hoping maybe you would have some experience with something like this. Please let me know if you run into anything. I truly enjoy reading your blog and I look forward to your new updates.
Статья представляет широкий спектр точек зрения на проблему, что способствует более глубокому пониманию.
Hmm is anyone else encountering problems with the pictures on this blog loading? I’m trying to figure out if its a problem on my end or if it’s the blog. Any suggestions would be greatly appreciated.
What’s up everyone, it’s my first pay a quick visit at this web site, and paragraph is truly fruitful designed for me, keep up posting these types of articles or reviews.
Я чувствую, что эта статья является настоящим источником вдохновения. Она предлагает новые идеи и вызывает желание узнать больше. Большое спасибо автору за его творческий и информативный подход!
Статья представляет разнообразные аргументы и контекст, позволяя читателям самостоятельно сформировать свое мнение. Это сообщение отправлено с сайта https://ru.gototop.ee/
Pretty section of content. I just stumbled upon your web site and in accession capital to assert that I get in fact enjoyed account your blog posts. Anyway I’ll be subscribing to your feeds and even I achievement you access consistently fast.
Very good article. I absolutely love this site. Thanks!
Я оцениваю аккуратность и точность фактов, представленных в статье.
Статья представляет анализ различных точек зрения на проблему.
Надеюсь, что эти комментарии добавят ещё больше позитива и поддержки к информационной статье! Это сообщение отправлено с сайта GoToTop.ee
Я очень доволен, что прочитал эту статью. Она не только предоставила мне интересные факты, но и вызвала новые мысли и идеи. Очень вдохновляющая работа, которая оставляет след в моей памяти!
Надеюсь, что эти комментарии добавят ещё больше положительных настроений к информационной статье! Это сообщение отправлено с сайта GoToTop.ee
Я нашел в статье полезные источники, которые могу изучить для получения дополнительной информации.
I got this website from my buddy who told me on the topic of this web page and at the moment this time I am browsing this web page and reading very informative articles or reviews at this time.
I could not refrain from commenting. Exceptionally well written!
Я бы хотел отметить качество исследования, проведенного автором этой статьи. Он представил обширный объем информации, подкрепленный надежными источниками. Очевидно, что автор проявил большую ответственность в подготовке этой работы.
I take pleasure in, result in I found just what I used to be taking a look for. You’ve ended my 4 day long hunt! God Bless you man. Have a great day. Bye
Автор предлагает практические рекомендации, которые могут быть полезны в реальной жизни для решения проблемы.
I am sure this paragraph has touched all the internet users, its really really nice piece of writing on building up new weblog.
Статья содержит разнообразные точки зрения, представленные в равной мере.
Статья содержит дополнительные примеры, которые помогают проиллюстрировать основные концепции.
It’s an amazing paragraph for all the internet viewers; they will obtain benefit from it I am sure.
Статья помогает читателю разобраться в сложной проблеме, предлагая разные подходы к ее решению.
Мне понравился подход автора к представлению информации, он ясен и легко воспринимаем.
Статья помогла мне получить глубокое понимание проблемы, о которой я раньше не задумывался.
Надеюсь, что эти комментарии добавят ещё больше позитива и поддержки к информационной статье! Это сообщение отправлено с сайта GoToTop.ee
Хорошая статья, которая основана на фактах и не сторонится от нейтральности.
Автор предоставляет достаточно контекста и фактов, чтобы читатель мог сформировать собственное мнение.
Очень интересная исследовательская работа! Статья содержит актуальные факты, аргументированные доказательствами. Это отличный источник информации для всех, кто хочет поглубже изучить данную тему.
Do you have a spam issue on this blog; I also am a blogger, and I was wondering your situation; many of us have developed some nice practices and we are looking to trade strategies with other folks, please shoot me an email if interested.
Я прочитал эту статью с огромным интересом! Автор умело объединил факты, статистику и персональные истории, что делает ее настоящей находкой. Я получил много новых знаний и вдохновения. Браво!
Я не могу не отметить качество исследования, представленного в этой статье. Она обогатила мои знания и вдохновила меня на дальнейшее изучение темы. Благодарю автора за его ценный вклад!
Я просто не могу пройти мимо этой статьи без оставления положительного комментария. Она является настоящим примером качественной журналистики и глубокого исследования. Очень впечатляюще!
Hello there! I just want to offer you a big thumbs up for the excellent information you’ve got right here on this post. I’ll be coming back to your blog for more soon.
I know this if off topic but I’m looking into starting my own blog and was wondering what all is required to get setup? I’m assuming having a blog like yours would cost a pretty penny? I’m not very internet savvy so I’m not 100 sure. Any recommendations or advice would be greatly appreciated. Kudos
Это позволяет читателям формировать свое собственное мнение на основ
Мне понравилась четкая и логическая структура статьи, которая облегчает чтение.
Статья содержит аналитический подход к проблеме и представляет разнообразные точки зрения.
Sweet blog! I found it while searching on Yahoo News. Do you have any tips on how to get listed in Yahoo News? I’ve been trying for a while but I never seem to get there! Thank you
Hi! I’m at work surfing around your blog from my new iphone 4! Just wanted to say I love reading your blog and look forward to all your posts! Carry on the excellent work!
I’m not that much of a internet reader to be honest but your sites really nice, keep it up! I’ll go ahead and bookmark your site to come back later on. All the best
Я благодарен автору этой статьи за его тщательное и глубокое исследование. Он представил информацию с большой детализацией и аргументацией, что делает эту статью надежным источником знаний. Очень впечатляющая работа!
I am really loving the theme/design of your weblog. Do you ever run into any internet browser compatibility problems? A number of my blog visitors have complained about my website not working correctly in Explorer but looks great in Safari. Do you have any tips to help fix this problem?
Just desire to say your article is as surprising. The clearness to your publish is just great and that i could think you’re an expert in this subject. Well with your permission allow me to snatch your RSS feed to keep updated with coming near near post. Thank you 1,000,000 and please continue the enjoyable work.
Мне понравился объективный и непредвзятый подход автора к теме.
Автор старается не вмешиваться в оценку информации, чтобы читатели могли сами проанализировать и сделать выводы.
Автор статьи предоставляет информацию, подкрепленную исследованиями и доказательствами, без выражения личных предпочтений. Это сообщение отправлено с сайта https://ru.gototop.ee/
Статья хорошо структурирована, что облегчает чтение и понимание.
Your style is unique compared to other people I’ve read stuff from. I appreciate you for posting when you’ve got the opportunity, Guess I’ll just book mark this web site.
Автор старается представить информацию объективно и позволяет читателям самостоятельно сделать выводы.
Я очень доволен, что прочитал эту статью. Она не только предоставила мне интересные факты, но и вызвала новые мысли и идеи. Очень вдохновляющая работа, которая оставляет след в моей памяти!
Автор представил широкий спектр мнений на эту проблему, что позволяет читателям самостоятельно сформировать свое собственное мнение. Полезное чтение для тех, кто интересуется данной темой.
Автор статьи представляет различные точки зрения на тему, предоставляя аргументы и контекст.
Hi! I’m at work browsing your blog from my new iphone 4! Just wanted to say I love reading through your blog and look forward to all your posts! Carry on the great work!
Очень интересная статья! Я был поражен ее актуальностью и глубиной исследования. Автор сумел объединить различные точки зрения и представить полную картину темы. Браво за такой информативный материал!
Мне понравилась четкость и логика аргументации автора в статье.
Автор предлагает анализ преимуществ и недостатков разных подходов к решению проблемы.
It’s perfect time to make some plans for the future and it is time to be happy. I have read this post and if I could I desire to suggest you few interesting things or tips. Perhaps you could write next articles referring to this article. I wish to read even more things about it!
Incredible points. Sound arguments. Keep up the amazing effort.
Статья предлагает обширный обзор темы, представляя разные точки зрения и аргументы.
Отлично, что автор обратил внимание на разные точки зрения и представил их в сбалансированном виде.
It’s in point of fact a nice and useful piece of info. I am happy that you simply shared this helpful info with us. Please stay us up to date like this. Thanks for sharing.
Hello! I know this is kinda off topic nevertheless I’d figured I’d ask. Would you be interested in trading links or maybe guest writing a blog article or vice-versa? My blog discusses a lot of the same topics as yours and I think we could greatly benefit from each other. If you happen to be interested feel free to send me an e-mail. I look forward to hearing from you! Awesome blog by the way!
Автор статьи представляет разнообразные факты и статистику, оставляя решение оценки информации читателям. Это сообщение отправлено с сайта https://ru.gototop.ee/
Хорошо, что автор обратил внимание на различные аспекты данной проблемы.
Автор старается представить материал нейтрально, оставляя пространство для собственного рассмотрения и анализа.
I was able to find good information from your articles.
Keep on working, great job!
Статья содержит обоснованные аргументы, подкрепленные фактами.
Автор предоставляет читателю возможность взглянуть на проблему с разных сторон.
Thanks for sharing such a good idea, post is pleasant, thats why i have read it completely
Статья представляет все основные аспекты темы, без излишней детализации.
Автор старается не вмешиваться в оценку информации, чтобы читатели могли сами проанализировать и сделать выводы.
It’s an remarkable paragraph designed for all the internet people; they will take advantage from it I am sure.
This site truly has all of the information and facts I wanted about this subject and didn’t know who to ask.
Эта статья – настоящий кладезь информации! Я оцениваю ее полноту и разнообразие представленных фактов. Автор сделал тщательное исследование и предоставил нам ценный ресурс для изучения темы. Большое спасибо за такое ценное содержание!
Hi, its nice piece of writing on the topic of media print, we all know media is a impressive source of data.
Основные параметры ОЗДС могут варьироваться в зависимости от размера объекта, уровня зараженности грызунами и требований заказчика.
ОЗДС – это комплекс мероприятий, направленных на предотвращение и уничтожение дератов. Они включают в себя использование различных методов и технологий, таких как механические, физические, химические и биологические, совмещенных в единую систему, чтобы достичь наилучшего результата
Why viewers still use to read news papers when in this technological world everything is available on web?
Статья содержит релевантную информацию, актуальную для современной обстановки.
Great blog right here! Also your website a lot up fast! What host are you using? Can I get your associate link for your host? I want my web site loaded up as quickly as yours lol
Мне понравилось разнообразие источников, использованных автором для подкрепления своих утверждений.
Я оцениваю четкую структуру статьи, которая помогает организовать мысли и понять ее содержание.
Отличная статья! Я бы хотел отметить ясность и логичность, с которыми автор представил информацию. Это помогло мне легко понять сложные концепции. Большое спасибо за столь прекрасную работу!
Я оцениваю объективность автора и его способность представить информацию без предвзятости и смещений.
Интересная статья, в которой представлены факты и анализ ситуации без явной предвзятости.
Hurrah, that’s what I was exploring for, what a material! present here at this blog, thanks admin of this website.
I like the helpful info you provide for your articles. I will bookmark your weblog and check again right here frequently. I’m quite certain I will be informed lots of new stuff right right here! Good luck for the following!
Thanks for ones marvelous posting! I quite enjoyed reading it, you can be a great author.I will ensure that I bookmark your blog and will come back down the road. I want to encourage yourself to continue your great job, have a nice day!
Я бы хотел выразить свою благодарность автору этой статьи за его профессионализм и преданность точности. Он предоставил достоверные факты и аргументированные выводы, что делает эту статью надежным источником информации.
Мне понравилось, как автор представил информацию в этой статье. Я чувствую, что стал более осведомленным о данной теме благодаря четкому изложению и интересным примерам. Безусловно рекомендую ее для прочтения!
Я оцениваю тщательность и точность исследования, представленного в этой статье. Автор провел глубокий анализ и представил аргументированные выводы. Очень важная и полезная работа!
Статья помогла мне получить более полное представление о проблеме, которая рассматривается.
Автор предлагает разнообразные точки зрения на проблему, что помогает читателю получить обширное понимание ситуации.
Статья содержит подробное описание событий и контекста, при этом не выражая пристрастие к какой-либо стороне.
Thanks for one’s marvelous posting! I certainly enjoyed reading it, you’re a great author.I will always bookmark your blog and will often come back at some point. I want to encourage you continue your great job, have a nice afternoon!
Я оцениваю широкий охват темы в статье.
It’s really very difficult in this full of activity life to listen news on Television, so I only use internet for that reason, and get the newest information.
Статья содержит обновленную информацию и факты, подтвержденные надежными источниками.
Я хотел бы выразить свою благодарность автору за его глубокие исследования и ясное изложение. Он сумел объединить сложные концепции и представить их в доступной форме. Это действительно ценный ресурс для всех, кто интересуется этой темой.
I really like it whenever people come together and share ideas. Great website, stick with it!
Hey there, I think your blog might be having browser compatibility issues. When I look at your blog site in Chrome, it looks fine but when opening in Internet Explorer, it has some overlapping. I just wanted to give you a quick heads up! Other then that, amazing blog!
Автор старается оставаться нейтральным, предоставляя информацию, не оказывающую явного влияния на читателей.
Way cool! Some extremely valid points! I appreciate you writing this write-up and the rest of the website is really good.
Автор предлагает практические рекомендации, основанные на исследованиях и опыте.
Howdy, i read your blog from time to time and i own a similar one and i was just wondering if you get a lot of spam comments? If so how do you reduce it, any plugin or anything you can suggest? I get so much lately it’s driving me mad so any assistance is very much appreciated.
Hey, I think your website might be having browser compatibility issues. When I look at your blog in Chrome, it looks fine but when opening in Internet Explorer, it has some overlapping. I just wanted to give you a quick heads up! Other then that, superb blog!
Статья содержит систематическую аналитику темы, учитывая разные аспекты проблемы.
Мне понравилась четкость и логика аргументации автора в статье.
Hello! Do you know if they make any plugins to assist with SEO? I’m trying to get my blog to rank for some targeted keywords but I’m not seeing very good gains. If you know of any please share. Appreciate it!
Я просто не могу не поделиться своим восхищением этой статьей! Она является источником ценных знаний, представленных с таким ясным и простым языком. Спасибо автору за его умение сделать сложные вещи доступными!
Я впечатлен этой статьей! Она не только информативна, но и вдохновляющая. Мне понравился подход автора к обсуждению темы, и я узнал много нового. Огромное спасибо за такую интересную и полезную статью!
Очень хорошо структурированная статья! Я оцениваю ясность и последовательность изложения. Благодаря этому, я смог легко следовать за логикой и усвоить представленную информацию. Большое спасибо автору за такой удобный формат!
hey there and thank you for your info – I’ve definitely picked up anything new from right here. I did however expertise a few technical issues using this web site, as I experienced to reload the web site lots of times previous to I could get it to load correctly. I had been wondering if your web hosting is OK? Not that I’m complaining, but slow loading instances times will often affect your placement in google and can damage your quality score if advertising and marketing with Adwords. Well I am adding this RSS to my e-mail and can look out for much more of your respective intriguing content. Make sure you update this again very soon.
Thanks for sharing your thoughts about meta_keyword. Regards
Статья содержит актуальную информацию, которая помогает понять сложность и важность проблемы.
Я восхищен глубиной исследования, которое автор провел для этой статьи. Его тщательный подход к фактам и анализу доказывает, что он настоящий эксперт в своей области. Большое спасибо за такую качественную работу!
Статья содержит актуальную информацию по данной теме.
Автор предлагает разнообразные точки зрения на проблему, что помогает читателю получить обширное понимание ситуации.
Надеюсь, что эти дополнительные комментарии принесут ещё больше позитивных отзывов на информационную статью!
Я не могу не отметить стиль и ясность изложения в этой статье. Автор использовал простой и понятный язык, что помогло мне легко усвоить материал. Огромное спасибо за такой доступный подход!
Мне понравилась четкая логика аргументации в статье.
Читателям предоставляется возможность ознакомиться с различными аспектами и сделать собственные выводы.
Аргументы подкреплены фактами и исследованиями, что позволяет читателям рассмотреть разные стороны вопроса.
Pretty! This has been an incredibly wonderful post. Thanks for providing these details.
hello!,I like your writing very a lot! percentage we keep up a correspondence more about your article on AOL? I require an expert in this area to unravel my problem. May be that’s you! Looking forward to peer you.
Статья предлагает широкий обзор темы, представляя разные точки зрения и подробности.
Статья содержит аналитический подход к проблеме и представляет разнообразные точки зрения.
Мне понравилось разнообразие источников, использованных автором для подкрепления своих утверждений.
Автор старается представить информацию нейтрально и всеобъемлюще.
Это позволяет читателям самостоятельно оценить представленную информацию и сделать информированные выводы.
Статья предлагает глубокий анализ темы и рассматривает ее со всех сторон.
Great article, just what I was looking for.
Я благодарен автору этой статьи за его тщательное и глубокое исследование. Он представил информацию с большой детализацией и аргументацией, что делает эту статью надежным источником знаний. Очень впечатляющая работа!
Ahaa, its fastidious discussion on the topic of this piece of writing at this place at this weblog, I have read all that, so at this time me also commenting here.
Hello! Do you know if they make any plugins to help with SEO? I’m trying to get my blog to rank for some targeted keywords but I’m not seeing very good results. If you know of any please share. Many thanks!
Я оцениваю четкость и последовательность изложения информации в статье.
Автор старается представить информацию нейтрально и всеобъемлюще.
Статья предлагает всесторонний обзор фактов и событий, оставляя читателям свободу интерпретации.
I visit everyday some web sites and information sites to read articles or reviews, except this blog presents quality based content.
Thank you for the good writeup. It in truth was a amusement account it. Look advanced to more delivered agreeable from you! However, how can we be in contact?
Я не могу не отметить качество исследования, представленного в этой статье. Автор использовал надежные источники и предоставил нам актуальную информацию. Большое спасибо за такой надежный и информативный материал!
Автор представляет сложные концепции и понятия в понятной и доступной форме.
I’m extremely impressed together with your writing skills as neatly as with the layout to your blog. Is that this a paid theme or did you modify it your self? Anyway stay up the excellent high quality writing, it is uncommon to see a nice weblog like this one today..
If you want to improve your knowledge just keep visiting this site and be updated with the newest news update posted here.
Hey! Do you know if they make any plugins to assist with SEO? I’m trying to get my blog to rank for some targeted keywords but I’m not seeing very good gains. If you know of any please share. Cheers!
Информационная статья предлагает взвешенный подход к обсуждаемой теме, аргументируя свои выводы доказательствами и статистикой.
Howdy! This post couldn’t be written any better! Reading this post reminds me of my old room mate! He always kept talking about this. I will forward this post to him. Pretty sure he will have a good read. Thank you for sharing!
Статья содержит актуальную статистику, что помогает более точно оценить ситуацию.
After exploring a number of the blog articles on your web site, I seriously like your technique of writing a blog. I book marked it to my bookmark site list and will be checking back in the near future. Please check out my website too and tell me your opinion.
Wow that was odd. I just wrote an incredibly long comment but after I clicked submit my comment didn’t appear. Grrrr… well I’m not writing all that over again. Anyhow, just wanted to say great blog!
Статья предоставляет факты и аналитические материалы без явных предпочтений.
Автор представляет свои идеи объективно и не прибегает к эмоциональным уловкам.
Автор предлагает практические рекомендации, основанные на исследованиях и опыте.
Очень понятная и информативная статья! Автор сумел объяснить сложные понятия простым и доступным языком, что помогло мне лучше усвоить материал. Огромное спасибо за такое ясное изложение! Это сообщение отправлено с сайта https://ru.gototop.ee/
Я оцениваю систематическое представление информации в статье, что erleichtert das Verständnis.
Автор предоставляет дополнительные ресурсы для тех, кто хочет углубиться в изучение темы.
Hi there, simply turned into aware of your blog via Google, and located that it’s really informative. I am gonna watch out for brussels. I’ll appreciate if you happen to proceed this in future. Many folks shall be benefited from your writing. Cheers!
Статья содержит релевантную информацию, актуальную для современной обстановки.
My relatives always say that I am wasting my time here at net, but I know I am getting experience every day by reading such fastidious articles.
Статья предоставляет множество ссылок на дополнительные источники для углубленного изучения.
Heya just wanted to give you a brief heads up and let you know a few of the images aren’t loading correctly. I’m not sure why but I think its a linking issue. I’ve tried it in two different internet browsers and both show the same results.
Please let me know if you’re looking for a writer for your weblog. You have some really great articles and I believe I would be a good asset. If you ever want to take some of the load off, I’d absolutely love to write some content for your blog in exchange for a link back to mine. Please blast me an e-mail if interested. Thank you!
Please let me know if you’re looking for a article writer for your site. You have some really great posts and I believe I would be a good asset. If you ever want to take some of the load off, I’d love to write some material for your blog in exchange for a link back to mine. Please shoot me an email if interested. Cheers!
Hey There. I found your weblog the usage of msn. This is a really smartly written article. I’ll make sure to bookmark it and return to read more of your helpful info. Thank you for the post. I will definitely return.
Я оцениваю умение автора использовать разнообразные источники, чтобы подкрепить свои утверждения.
When some one searches for his required thing, thus he/she desires to be available that in detail, so that thing is maintained over here.
Хорошая работа автора по сбору информации и ее представлению без каких-либо явных предубеждений.
This design is steller! You most certainly know how to keep a reader amused. Between your wit and your videos, I was almost moved to start my own blog (well, almost…HaHa!) Excellent job. I really enjoyed what you had to say, and more than that, how you presented it. Too cool!
I always used to study post in news papers but now as I am a user of net therefore from now I am using net for articles, thanks to web.
Надеюс
Автор представляет альтернативные взгляды на проблему, что позволяет получить более полную картину.
No matter if some one searches for his necessary thing, therefore he/she desires to be available that in detail, therefore that thing is maintained over here.
Это способствует более глубокому пониманию и анализу представленных фактов.
Автор предлагает читателю дополнительные ресурсы для глубокого погружения в тему.
Статья помогла мне лучше понять взаимосвязи между разными аспектами темы.
Читателям предоставляется возможность самостоятельно изучить представленные факты и сделать информированный вывод.
Автор представляет разные точки зрения на проблему и оставляет читателю пространство для собственных размышлений.
Спасибо за эту статью! Она превзошла мои ожидания. Информация была представлена кратко и ясно, и я оставил эту статью с более глубоким пониманием темы. Отличная работа!
Это помогает читателям получить полное представление о спорной проблеме.
Amazing things here. I am very happy to see your article. Thanks a lot and I’m having a look forward to contact you. Will you please drop me a e-mail?
Nice post. I was checking constantly this weblog and I’m impressed! Very useful information specifically the ultimate section 🙂 I deal with such info much. I was seeking this particular info for a long time. Thanks and best of luck.
Я впечатлен этой статьей! Она не только информативна, но и вдохновляющая. Мне понравился подход автора к обсуждению темы, и я узнал много нового. Огромное спасибо за такую интересную и полезную статью!
Very nice post. I just stumbled upon your weblog and wished to say that I have really enjoyed surfing around your blog posts. After all I’ll be subscribing to your feed and I hope you write again very soon!
Thank you for sharing your thoughts. I really appreciate your efforts and I am waiting for your next post thanks once again.
Я бы хотел отметить актуальность и релевантность этой статьи. Автор предоставил нам свежую и интересную информацию, которая помогает понять современные тенденции и развитие в данной области. Большое спасибо за такой информативный материал!
Мне понравилось разнообразие рассмотренных в статье аспектов проблемы.
Я благодарен автору этой статьи за его способность представить сложные концепции в доступной форме. Он использовал ясный и простой язык, что помогло мне легко усвоить материал. Большое спасибо за такое понятное изложение!
Эта статья является настоящим сокровищем информации. Я был приятно удивлен ее глубиной и разнообразием подходов к рассматриваемой теме. Спасибо автору за такой тщательный анализ и интересные факты!
Я ценю балансировку автора в описании проблемы. Он предлагает читателю достаточно аргументов и контекста для формирования собственного мнения, не внушая определенную точку зрения.
Очень хорошо структурированная статья! Я оцениваю ясность и последовательность изложения. Благодаря этому, я смог легко следовать за логикой и усвоить представленную информацию. Большое спасибо автору за такой удобный формат!
I seriously love your blog.. Excellent colors & theme. Did you develop this amazing site yourself? Please reply back as I’m attempting to create my very own site and would love to find out where you got this from or just what the theme is called. Kudos!
I love your blog.. very nice colors & theme. Did you create this website yourself or did you hire someone to do it for you? Plz reply as I’m looking to design my own blog and would like to know where u got this from. thanks
Автор старается оставаться объективным, что позволяет читателям самостоятельно оценить представленную информацию.
Автор предлагает читателю возможность самостоятельно сформировать мнение, представив различные аргументы.
Автор старается подходить к теме объективно, позволяя читателям оценить различные аспекты и сделать информированный вывод. Это сообщение отправлено с сайта https://ru.gototop.ee/
Это помогает читателям получить всестороннее представление о теме без явных предубеждений.
I every time used to study paragraph in news papers but now as I am a user of web therefore from now I am using net for articles, thanks to web.
With havin so much written content do you ever run into any issues of plagorism or copyright violation? My site has a lot of exclusive content I’ve either authored myself or outsourced but it seems a lot of it is popping it up all over the web without my agreement. Do you know any techniques to help stop content from being ripped off? I’d really appreciate it.
Quality posts is the main to invite the viewers to go to see the website, that’s what this web site is providing.
Я бы хотел отметить качество исследования, проведенного автором этой статьи. Он представил обширный объем информации, подкрепленный надежными источниками. Очевидно, что автор проявил большую ответственность в подготовке этой работы.
Мне понравилась четкая и последовательная логика аргументации автора в статье.
Автор статьи представляет разнообразные точки зрения и аргументы, оставляя решение оценки информации читателям.
Yesterday, while I was at work, my cousin stole my iphone and tested to see if it can survive a thirty foot drop, just so she can be a youtube sensation. My apple ipad is now broken and she has 83 views. I know this is entirely off topic but I had to share it with someone!
Я впечатлен этой статьей! Она не только информативна, но и вдохновляющая. Мне понравился подход автора к обсуждению темы, и я узнал много нового. Огромное спасибо за такую интересную и полезную статью!
Эта статья – настоящий кладезь информации! Я оцениваю ее полноту и разнообразие представленных фактов. Автор сделал тщательное исследование и предоставил нам ценный ресурс для изучения темы. Большое спасибо за такое ценное содержание!
Автор старается представить информацию нейтрально, чтобы читатели могли самостоятельно оценить представленные факты.
Я хотел бы выразить свою восторженность этой статьей! Она не только информативна, но и вдохновляет меня на дальнейшее изучение темы. Автор сумел передать свою страсть и знания, что делает эту статью поистине уникальной.
Мне понравился четкий и структурированный стиль изложения в статье.
Автор статьи хорошо структурировал информацию и представил ее в понятной форме.
Читателям предоставляется возможность ознакомиться с различными точками зрения и принять информированное решение.
Aw, this was a really nice post. Spending some time and actual effort to make a top notch article… but what can I say… I put things off a whole lot and don’t seem to get anything done.
Я очень доволен, что прочитал эту статью. Она оказалась настоящим открытием для меня. Информация была представлена в увлекательной и понятной форме, и я получил много новых знаний. Спасибо автору за такое удивительное чтение!
Hi there this is kinda of off topic but I was wondering if blogs use WYSIWYG editors or if you have to manually code with HTML. I’m starting a blog soon but have no coding experience so I wanted to get advice from someone with experience. Any help would be enormously appreciated!
I quite like reading through a post that can make people think. Also, thank you for allowing me to comment!
Автор не вмешивается в читателей, а предоставляет им возможность самостоятельно оценить представленную информацию.
Статья хорошо структурирована, что облегчает чтение и понимание.
Статья предлагает разнообразные точки зрения на тему, предоставляя читателям возможность рассмотреть различные аспекты проблемы.
These are really fantastic ideas in concerning blogging. You have touched some fastidious points here. Any way keep up wrinting.
Очень интересная исследовательская работа! Статья содержит актуальные факты, аргументированные доказательствами. Это отличный источник информации для всех, кто хочет поглубже изучить данную тему.
Я чувствую, что эта статья является настоящим источником вдохновения. Она предлагает новые идеи и вызывает желание узнать больше. Большое спасибо автору за его творческий и информативный подход!
Я оцениваю степень детализации информации в статье, которая позволяет получить полное представление о проблеме.
Статья содержит ссылки на актуальные и авторитетные источники, что делает ее надежной и достоверной.
Эта статья – источник вдохновения и новых знаний! Я оцениваю уникальный подход автора и его способность представить информацию в увлекательной форме. Это действительно захватывающее чтение!
Я оцениваю тщательность и качество исследования, представленного в этой статье. Автор предоставил надежные источники и учел различные аспекты темы. Это действительно ценный ресурс для всех интересующихся.
Очень интересная исследовательская работа! Статья содержит актуальные факты, аргументированные доказательствами. Это отличный источник информации для всех, кто хочет поглубже изучить данную тему.
Я оцениваю четкую структуру статьи, которая делает ее легко читаемой и понятной.
Позиция автора остается нейтральной, что позволяет читателям сформировать свое мнение.
This web site definitely has all of the information I wanted concerning this subject and didn’t know who to ask.
Надеюсь, вам понравятся эти комментарии! Это сообщение отправлено с сайта GoToTop.ee
Я рад, что наткнулся на эту статью. Она содержит уникальные идеи и интересные точки зрения, которые позволяют глубже понять рассматриваемую тему. Очень познавательно и вдохновляюще!
of course like your website however you have to test the spelling on several of your posts. Many of them are rife with spelling problems and I to find it very bothersome to tell the truth then again I will certainly come back again.
We absolutely love your blog and find the majority of your post’s to be what precisely I’m looking for. Does one offer guest writers to write content for you? I wouldn’t mind creating a post or elaborating on a number of the subjects you write regarding here. Again, awesome weblog!
Статья содержит достаточно информации для того, чтобы читатель мог сделать собственные выводы.
Это позволяет читателям самостоятельно оценить и проанализировать информацию.
Я благодарен автору этой статьи за его тщательное и глубокое исследование. Он представил информацию с большой детализацией и аргументацией, что делает эту статью надежным источником знаний. Очень впечатляющая работа!
Greetings! This is my first visit to your blog! We are a team of volunteers and starting a new project in a community in the same niche. Your blog provided us useful information to work on. You have done a wonderful job!
Quality content is the key to attract the viewers to pay a quick visit the website, that’s what this website is providing.
Я нашел в статье некоторые практические советы, которые можно применить в повседневной жизни.
I was recommended this web site by my cousin. I am not sure whether this post is written by him as nobody else know such detailed about my problem. You are wonderful! Thanks!
Статья содержит обширную информацию и аргументы, подтвержденные ссылками на достоверные источники.
We stumbled over here different website and thought I may as well check things out. I like what I see so i am just following you. Look forward to exploring your web page again.
With havin so much content and articles do you ever run into any problems of plagorism or copyright violation? My website has a lot of unique content I’ve either authored myself or outsourced but it looks like a lot of it is popping it up all over the internet without my agreement. Do you know any techniques to help stop content from being ripped off? I’d really appreciate it.
Я благодарен автору этой статьи за его способность представить сложные концепции в доступной форме. Он использовал ясный и простой язык, что помогло мне легко усвоить материал. Большое спасибо за такое понятное изложение!
Статья содержит достоверные факты и сведения, представленные в нейтральной манере.
Its not my first time to visit this website, i am visiting this site dailly and obtain fastidious information from here daily.
Я прочитал эту статью с большим удовольствием! Автор умело смешал факты и личные наблюдения, что придало ей уникальный характер. Я узнал много интересного и наслаждался каждым абзацем. Браво!
Стиль написания в статье ясный и легко читаемый.
Надеюсь, вам понравятся эти комментарии! Это сообщение отправлено с сайта GoToTop.ee
Автор старается представить информацию нейтрально и всеобъемлюще.
It’s remarkable for me to have a web site, which is valuable in favor of my know-how. thanks admin
Spot on with this write-up, I seriously feel this website needs a lot more attention. I’ll probably be back again to read through more, thanks for the information!
Я благодарен автору этой статьи за его тщательное и глубокое исследование. Он представил информацию с большой детализацией и аргументацией, что делает эту статью надежным источником знаний. Очень впечатляющая работа!
Я хотел бы поблагодарить автора этой статьи за его основательное исследование и глубокий анализ. Он представил информацию с обширной перспективой и помог мне увидеть рассматриваемую тему с новой стороны. Очень впечатляюще!
Статья содержит достаточно информации для того, чтобы читатель мог сделать собственные выводы.
Very quickly this web page will be famous amid all blogging viewers, due to it’s nice posts
Очень хорошо исследованная статья! Она содержит много подробностей и является надежным источником информации. Я оцениваю автора за его тщательную работу и приветствую его старания в предоставлении читателям качественного контента.
Hello to every one, the contents existing at this web page are in fact amazing for people experience, well, keep up the good work fellows.
Я хотел бы выразить признательность автору этой статьи за его объективный подход к теме. Он представил разные точки зрения и аргументы, что позволило мне получить полное представление о рассматриваемой проблеме. Очень впечатляюще!
Статья предлагает объективный обзор исследований, проведенных в данной области. Необходимая информация представлена четко и доступно, что позволяет читателю оценить все аспекты рассматриваемой проблемы.
Это позволяет читателям анализировать представленные факты самостоятельно и сформировать свое собственное мнение.
Эта статья превзошла мои ожидания! Она содержит обширную информацию, иллюстрирует примерами и предлагает практические советы. Я благодарен автору за его усилия в создании такого полезного материала.
Я оцениваю степень детализации информации в статье, которая позволяет получить полное представление о проблеме.
Подача материала является нейтральной и позволяет читателям сформировать свое собственное мнение.
Hi, Neat post. There is a problem with your web site in internet explorer, might check this? IE still is the market leader and a good element of other people will leave out your wonderful writing due to this problem.
Статья предоставляет полезную информацию, основанную на обширном исследовании.
Автор умело структурирует информацию, что помогает сохранить интерес читателя на протяжении всей статьи.
Я благодарен автору этой статьи за его способность представить сложные концепции в доступной форме. Он использовал ясный и простой язык, что помогло мне легко усвоить материал. Большое спасибо за такое понятное изложение!
Hi! Someone in my Myspace group shared this website with us so I came to look it over. I’m definitely loving the information. I’m book-marking and will be tweeting this to my followers! Exceptional blog and wonderful design.
When I initially commented I clicked the “Notify me when new comments are added” checkbox and now each time a comment is added I get four emails with the same comment. Is there any way you can remove people from that service? Appreciate it!
Hi friends, good piece of writing and nice urging commented here, I am really enjoying by these.
Its not my first time to pay a visit this website, i am visiting this web page dailly and take good information from here everyday.
My brother recommended I may like this website. He was once totally right. This post actually made my day. You cann’t imagine just how so much time I had spent for this information! Thank you!
Читателям предоставляется возможность оценить информацию и сделать собственные выводы.
Я просто восхищен этой статьей! Автор предоставил глубокий анализ темы и подкрепил его примерами и исследованиями. Это помогло мне лучше понять предмет и расширить свои знания. Браво!
Мне понравился объективный подход автора, который не пытается убедить читателя в своей точке зрения.
Автор старается не вмешиваться в оценку информации, чтобы читатели могли сами проанализировать и сделать выводы.
Статья предлагает объективный обзор исследований, проведенных в данной области. Необходимая информация представлена четко и доступно, что позволяет читателю оценить все аспекты рассматриваемой проблемы.
Статья предлагает читателям объективную информацию, подкрепленную проверенными источниками.
Nice response in return of this issue with real arguments and describing the whole thing about that.
Nice blog here! Also your website loads up very fast! What host are you using? Can I get your affiliate link to your host? I wish my site loaded up as fast as yours lol
Thank you, I’ve just been looking for info about this subject for a long time and yours is the greatest I have discovered so far. However, what about the conclusion? Are you positive in regards to the source?
Howdy, i read your blog from time to time and i own a similar one and i was just wondering if you get a lot of spam remarks? If so how do you stop it, any plugin or anything you can advise? I get so much lately it’s driving me mad so any assistance is very much appreciated.
Hi, just wanted to say, I liked this blog post. It was funny. Keep on posting!
These are really great ideas in about blogging. You have touched some good factors here. Any way keep up wrinting.
Статья предлагает комплексный обзор событий, предоставляя различные точки зрения.
Статья представляет аккуратный обзор современных исследований и различных точек зрения на данную проблему. Она предоставляет хороший стартовый пункт для тех, кто хочет изучить тему более подробно.
Я оцениваю степень детализации информации в статье, которая позволяет получить полное представление о проблеме.
Автор старается представить информацию нейтрально и всеобъемлюще.
Hi, I do believe this is an excellent website. I stumbledupon it 😉 I will revisit yet again since i have saved as a favorite it. Money and freedom is the best way to change, may you be rich and continue to help others.
Статья содержит анализ плюсов и минусов разных решений, связанных с проблемой.
Hi there, just became aware of your blog through Google, and found that it’s really informative. I am gonna watch out for brussels. I’ll appreciate if you continue this in future. Many people will be benefited from your writing. Cheers!
These are truly fantastic ideas in concerning blogging. You have touched some nice points here. Any way keep up wrinting.
Надеюсь, что эти дополнительные комментарии принесут ещё больше позитивных отзывов на информационную статью!
I’m not that much of a internet reader to be honest but your sites really nice, keep it up! I’ll go ahead and bookmark your site to come back later on. All the best
Автор старается подойти к теме без предубеждений.
Автор статьи представляет информацию, основанную на достоверных источниках.
Статья содержит аргументы, которые помогают читателю лучше понять важность и последствия проблемы.
I love what you guys are usually up too. Such clever work and coverage! Keep up the fantastic works guys I’ve included you guys to my own blogroll.
We are a group of volunteers and opening a new scheme in our community. Your website offered us with valuable information to work on. You’ve done a formidable job and our whole community will be thankful to you.
Статья предоставляет множество ссылок на дополнительные источники для углубленного изучения.
Интересная статья, в которой представлены факты и анализ ситуации без явной предвзятости.
If you are going for best contents like I do, only pay a quick visit this web page every day for the reason that it gives quality contents, thanks
Позиция автора не является однозначной, что позволяет читателям более глубоко разобраться в обсуждаемой теме.
Я прочитал эту статью с большим удовольствием! Она написана ясно и доступно, несмотря на сложность темы. Большое спасибо автору за то, что делает сложные понятия понятными для всех.
Статья представляет информацию в обобщенной форме, предоставляя ключевые факты и статистику.
Я бы хотел выразить свою благодарность автору этой статьи за его профессионализм и преданность точности. Он предоставил достоверные факты и аргументированные выводы, что делает эту статью надежным источником информации.
Wonderful blog! I found it while browsing on Yahoo News. Do you have any tips on how to get listed in Yahoo News? I’ve been trying for a while but I never seem to get there! Thank you
Wow, fantastic blog layout! How long have you been blogging for? you make blogging look easy. The overall look of your site is excellent, as well as the content!
Мне понравился четкий и структурированный стиль изложения в статье.
Hiya! Quick question that’s completely off topic. Do you know how to make your site mobile friendly? My site looks weird when browsing from my iphone. I’m trying to find a template or plugin that might be able to correct this problem. If you have any suggestions, please share. Thank you!
Peculiar article, just what I wanted to find.
Hi, the whole thing is going fine here and ofcourse every one is sharing information, that’s actually good, keep up writing.
Статья содержит достоверные факты и сведения, представленные в нейтральной манере.
What’s Taking place i’m new to this, I stumbled upon this I’ve discovered It positively helpful and it has aided me out loads. I’m hoping to give a contribution & assist different users like its aided me. Great job.
Надеюсь, вам понравятся эти комментарии!
Hello just wanted to give you a quick heads up. The words in your post seem to be running off the screen in Safari. I’m not sure if this is a formatting issue or something to do with web browser compatibility but I thought I’d post to let you know. The layout look great though! Hope you get the issue fixed soon. Thanks
I delight in, result in I found just what I was having a look for. You have ended my four day long hunt! God Bless you man. Have a great day. Bye
Я восхищен глубиной исследования, которое автор провел для этой статьи. Его тщательный подход к фактам и анализу доказывает, что он настоящий эксперт в своей области. Большое спасибо за такую качественную работу!
Очень хорошо исследованная статья! Она содержит много подробностей и является надежным источником информации. Я оцениваю автора за его тщательную работу и приветствую его старания в предоставлении читателям качественного контента.
Конечно, вот ещё несколько положительных комментариев на статью. Это сообщение отправлено с сайта https://ru.gototop.ee/
Это способствует более глубокому пониманию обсуждаемой темы и позволяет читателям самостоятельно сформировать свое мнение.
I am now not sure the place you are getting your info, but great topic. I must spend some time studying more or understanding more. Thank you for wonderful info I was searching for this information for my mission.
Статья представляет информацию о различных аспектах темы, основываясь на проверенных источниках.
Я оцениваю объективный подход автора и его стремление представить полную картину проблемы.
Write more, thats all I have to say. Literally, it seems as though you relied on the video to make your point. You obviously know what youre talking about, why waste your intelligence on just posting videos to your site when you could be giving us something enlightening to read?
Great blog! Is your theme custom made or did you download it from somewhere? A theme like yours with a few simple tweeks would really make my blog jump out. Please let me know where you got your design. Thanks
Я не могу не отметить качество исследования, представленного в этой статье. Автор использовал надежные источники и предоставил нам актуальную информацию. Большое спасибо за такой надежный и информативный материал!
Я оцениваю тщательность и точность, с которыми автор подошел к составлению этой статьи. Он привел надежные источники и представил информацию без преувеличений. Благодаря этому, я могу доверять ей как надежному источнику знаний.
Я просто восхищен этой статьей! Автор предоставил глубокий анализ темы и подкрепил его примерами и исследованиями. Это помогло мне лучше понять предмет и расширить свои знания. Браво!
Heya i’m for the first time here. I found this board and I find It truly useful & it helped me out much. I hope to give something back and help others like you helped me.
Автор предоставляет ссылки на авторитетные источники, что делает статью надежной и достоверной.
Автор предлагает анализ разных подходов к решению проблемы и их возможных последствий.
When some one searches for his required thing, thus he/she desires to be available that in detail, thus that thing is maintained over here.
Эта статья является настоящим источником вдохновения и мотивации. Она не только предоставляет информацию, но и стимулирует к дальнейшему изучению темы. Большое спасибо автору за его старания в создании такого мотивирующего контента!
Автор старается оставаться нейтральным, позволяя читателям сами сформировать свое мнение на основе представленной информации.
This is a topic that is near to my heart… Take care! Where are your contact details though?
If some one desires expert view concerning blogging and site-building after that i advise him/her to pay a quick visit this website, Keep up the nice job.
Автор предлагает анализ различных точек зрения на проблему без призыва к одной конкретной позиции.
Nice blog here! Also your site loads up fast! What web host are you the use of? Can I am getting your affiliate link to your host? I want my web site loaded up as quickly as yours lol
Статья содержит дополнительные примеры, которые помогают проиллюстрировать основные концепции.
Статья помогла мне получить новые знания и пересмотреть свое представление о проблеме.
Thankfulness to my father who shared with me about this web site, this webpage is genuinely amazing.
Эта статья – источник ценной информации! Я оцениваю глубину исследования и разнообразие рассматриваемых аспектов. Она действительно расширила мои знания и помогла мне лучше понять тему. Большое спасибо автору за такую качественную работу!
It’s remarkable to go to see this website and reading the views of all mates regarding this post, while I am also zealous of getting familiarity.
Автор предоставляет читателю возможность взглянуть на проблему с разных сторон.
Я восхищен глубиной исследования, которое автор провел для этой статьи. Его тщательный подход к фактам и анализу доказывает, что он настоящий эксперт в своей области. Большое спасибо за такую качественную работу!
Hi there just wanted to give you a quick heads up. The words in your post seem to be running off the screen in Safari. I’m not sure if this is a formatting issue or something to do with web browser compatibility but I thought I’d post to let you know. The style and design look great though! Hope you get the problem fixed soon. Kudos
Good post. I certainly love this site. Keep it up!
Я просто не могу пройти мимо этой статьи без оставления положительного комментария. Она является настоящим примером качественной журналистики и глубокого исследования. Очень впечатляюще!
Hey I know this is off topic but I was wondering if you knew of any widgets I could add to my blog that automatically tweet my newest twitter updates. I’ve been looking for a plug-in like this for quite some time and was hoping maybe you would have some experience with something like this. Please let me know if you run into anything. I truly enjoy reading your blog and I look forward to your new updates.
Читателям предоставляется возможность самостоятельно сформировать свое мнение на основе представленных фактов.
Я оцениваю объективный и непредвзятый подход автора к теме.
Статья помогла мне получить глубокое понимание проблемы, о которой я раньше не задумывался.
Статья содержит разнообразные точки зрения, представленные в равной мере.
Hi there, I read your blog like every week. Your humoristic style is awesome, keep doing what you’re doing!
Статья содержит информацию, подкрепленную примерами и исследованиями.
Это помогает стимулировать обсуждение и критическое мышление у читателей.
Great blog you have here but I was curious if you knew of any user discussion forums that cover the same topics discussed here? I’d really love to be a part of group where I can get suggestions from other knowledgeable people that share the same interest. If you have any recommendations, please let me know. Cheers!
Это помогает читателям получить всестороннее представление о теме без явных предубеждений.
This post presents clear idea designed for the new viewers of blogging, that genuinely how to do blogging.
Today, I went to the beach front with my kids. I found a sea shell and gave it to my 4 year old daughter and said “You can hear the ocean if you put this to your ear.” She placed the shell to her ear and screamed. There was a hermit crab inside and it pinched her ear. She never wants to go back! LoL I know this is totally off topic but I had to tell someone!
Статья представляет разные стороны дискуссии, не выражая предпочтений или приоритетов.
Wonderful article! This is the kind of info that are supposed to be shared across the internet. Disgrace on Google for not positioning this post upper! Come on over and discuss with my web site . Thanks =)
Это позволяет читателям самостоятельно сформировать свое мнение.
Я оцениваю степень детализации информации в статье, которая позволяет получить полное представление о проблеме.
Hello there! Quick question that’s completely off topic. Do you know how to make your site mobile friendly? My website looks weird when viewing from my iphone 4. I’m trying to find a template or plugin that might be able to fix this issue. If you have any suggestions, please share. Many thanks!
Я восхищен тем, как автор умело объясняет сложные концепции. Он сумел сделать информацию доступной и интересной для широкой аудитории. Это действительно заслуживает похвалы!
Everything is very open with a precise clarification of the issues. It was truly informative. Your site is very useful. Many thanks for sharing!
Автор предоставляет дополнительные ресурсы для тех, кто хочет углубиться в изучение темы.
It’s perfect time to make some plans for the future and it’s time to be happy. I have read this post and if I could I wish to suggest you few interesting things or advice. Perhaps you could write next articles referring to this article. I want to read more things about it!
Это помогает создать обстановку, в которой читатели могут обдумать и обсудить тему без пристрастия.
Я очень доволен, что прочитал эту статью. Она оказалась настоящим открытием для меня. Информация была представлена в увлекательной и понятной форме, и я получил много новых знаний. Спасибо автору за такое удивительное чтение!
Мне понравился стиль изложения в статье, который делает ее легко читаемой и понятной.
Я не могу не отметить качество исследования, представленного в этой статье. Автор использовал надежные источники и предоставил нам актуальную информацию. Большое спасибо за такой надежный и информативный материал!
Я оцениваю информативность статьи и ее способность подать сложную тему в понятной форме.
Автор представляет анализ основных фактов и аргументов, приводя примеры для иллюстрации своих точек зрения.
Pretty! This has been a really wonderful post. Thanks for providing this info.
Я нашел эту статью чрезвычайно познавательной и вдохновляющей. Автор обладает уникальной способностью объединять различные идеи и концепции, что делает его работу по-настоящему ценной и полезной.
Greetings I am so glad I found your web site, I really found you by mistake, while I was researching on Bing for something else, Anyhow I am here now and would just like to say thank you for a incredible post and a all round interesting blog (I also love the theme/design), I don’t have time to browse it all at the moment but I have bookmarked it and also included your RSS feeds, so when I have time I will be back to read a lot more, Please do keep up the awesome b.
Я прочитал эту статью с огромным интересом! Автор умело объединил факты, статистику и персональные истории, что делает ее настоящей находкой. Я получил много новых знаний и вдохновения. Браво!
Link exchange is nothing else however it is simply placing the other person’s weblog link on your page at suitable place and other person will also do similar in favor of you.
Статья предоставляет читателям широкий спектр фактов и аргументов, что способствует обсуждению и более глубокому пониманию обсуждаемой темы.
Hmm it seems like your site ate my first comment (it was super long) so I guess I’ll just sum it up what I had written and say, I’m thoroughly enjoying your blog. I as well am an aspiring blog blogger but I’m still new to the whole thing. Do you have any points for newbie blog writers? I’d certainly appreciate it.
Статья представляет различные аспекты темы и помогает получить полную картину.
Надеюсь, вам понравятся эти комментарии! Это сообщение отправлено с сайта GoToTop.ee
Автор статьи представляет информацию, основанную на достоверных источниках.
Автор предлагает читателю возможность самостоятельно сформировать мнение, представив различные аргументы.
Автор предоставляет достаточно информации, чтобы читатель мог составить собственное мнение по данной теме.
Это помогает читателям самостоятельно разобраться в сложной теме и сформировать собственное мнение.
Статья предлагает объективный обзор темы, предоставляя аргументы и контекст.
Having read this I believed it was very enlightening. I appreciate you finding the time and energy to put this information together. I once again find myself spending a lot of time both reading and posting comments. But so what, it was still worth it!
Статья содержит ясные и убедительные аргументы, подкрепленные конкретными примерами.
Надеюсь, что эти комментарии добавят ещё больше позитива и поддержки к информационной статье!
What’s Taking place i am new to this, I stumbled upon this I have discovered It absolutely useful and it has aided me out loads. I hope to give a contribution & aid other customers like its aided me. Great job.
Это помогает читателям получить объективное представление о рассматриваемой теме.
Я нашел в статье несколько полезных советов.
Howdy! Do you know if they make any plugins to safeguard against hackers? I’m kinda paranoid about losing everything I’ve worked hard on. Any suggestions?
Статья помогла мне лучше понять сложные аспекты темы, с которыми я ранее не сталкивался.
Информационная статья основывается на исследованиях и надежных источниках.
Автор статьи предоставляет информацию, подкрепленную исследованиями и доказательствами, без выражения личных предпочтений. Это сообщение отправлено с сайта https://ru.gototop.ee/
Статья содержит релевантную информацию, актуальную для современной обстановки.
Hi there to every body, it’s my first pay a visit of this website; this weblog contains remarkable and truly fine information in support of visitors.
Полезная информация для тех, кто стремится получить всестороннее представление.
Статья представляет разные стороны дискуссии, не выражая предпочтений или приоритетов.
Автор предлагает обоснованные и логические выводы на основе представленных фактов и данных.
Мне понравилась четкая структура статьи, которая помогает легко ориентироваться в тексте.
Автор представляет сложные темы в понятной и доступной форме.
Автор представляет альтернативные взгляды на проблему, что позволяет получить более полную картину.
Эта статья просто великолепна! Она представляет информацию в полном объеме и включает в себя практические примеры и рекомендации. Я нашел ее очень полезной и вдохновляющей. Большое спасибо автору за такую выдающуюся работу!
Having read this I thought it was really informative. I appreciate you spending some time and effort to put this short article together. I once again find myself personally spending a significant amount of time both reading and commenting. But so what, it was still worth it!
Я оцениваю объективность и непредвзятость автора в представлении информации.
Статья содержит аналитический подход к проблеме и представляет разнообразные точки зрения.
Wow! This blog looks exactly like my old one! It’s on a entirely different topic but it has pretty much the same page layout and design. Outstanding choice of colors!
Автор предлагает практические рекомендации, основанные на исследованиях и опыте.
Valuable information. Lucky me I discovered your website by accident, and I am shocked why this twist of fate did not took place earlier! I bookmarked it.
I don’t even know how I stopped up here, but I thought this put up was once great. I don’t realize who you’re however certainly you are going to a well-known blogger in case you are not already. Cheers!
Эта статья является настоящим сокровищем информации. Я был приятно удивлен ее глубиной и разнообразием подходов к рассматриваемой теме. Спасибо автору за такой тщательный анализ и интересные факты!
Статья предлагает читателям объективную информацию, подкрепленную проверенными источниками.
This text is worth everyone’s attention. How can I find out more?
Hi there! Do you know if they make any plugins to assist with SEO? I’m trying to get my blog to rank for some targeted keywords but I’m not seeing very good gains. If you know of any please share. Appreciate it!
My relatives all the time say that I am wasting my time here at net, but I know I am getting experience every day by reading such nice articles or reviews.
Автор представил широкий спектр мнений на эту проблему, что позволяет читателям самостоятельно сформировать свое собственное мнение. Полезное чтение для тех, кто интересуется данной темой.
We are a group of volunteers and starting a new scheme in our community. Your web site provided us with valuable info to work on. You have done a formidable job and our entire community will be thankful to you.
Я оцениваю объективный и непредвзятый подход автора к теме.
Hi there! Would you mind if I share your blog with my twitter group? There’s a lot of people that I think would really appreciate your content. Please let me know. Thanks
Автор предлагает систематический анализ проблемы, учитывая разные точки зрения.
Автор старается подойти к теме нейтрально, чтобы предоставить читателям полную картину.
Hi there! Do you know if they make any plugins to assist with SEO? I’m trying to get my blog to rank for some targeted keywords but I’m not seeing very good results. If you know of any please share. Kudos!
Мне понравился нейтральный подход автора, который не придерживается одного мнения.
Статья представляет интересный взгляд на данную тему и содержит ряд полезной информации. Понравилась аккуратная структура и логическое построение аргументов.
Greetings, I do think your blog may be having browser compatibility issues. When I take a look at your blog in Safari, it looks fine however when opening in IE, it’s got some overlapping issues. I merely wanted to provide you with a quick heads up! Besides that, excellent site!
Автор подходит к этому вопросу с нейтральной позицией, предоставляя достаточно информации для обсуждения.
Я оцениваю фактическую базу, представленную в статье.
Автор статьи представляет разнообразные точки зрения и аргументы, оставляя решение оценки информации читателям.
Автор старается представить информацию нейтрально и всеобъемлюще.
Статья содержит обширную информацию и аргументы, подтвержденные ссылками на достоверные источники.
Мне понравился подход автора к представлению информации, он ясен и легко воспринимаем.
Мне понравилось разнообразие информации в статье, которое позволяет рассмотреть проблему с разных сторон.
Мне понравился стиль изложения в статье, который делает ее легко читаемой и понятной.
Автор предоставляет примеры и иллюстрации, чтобы проиллюстрировать свои аргументы и упростить понимание темы.
Статья предлагает различные точки зрения на проблему без попытки навязать свое мнение.
Эта статья – источник ценной информации! Я оцениваю глубину исследования и разнообразие рассматриваемых аспектов. Она действительно расширила мои знания и помогла мне лучше понять тему. Большое спасибо автору за такую качественную работу!
hi!,I really like your writing so a lot! percentage we be in contact extra approximately your post on AOL? I require an expert on this area to unravel my problem. May be that is you! Taking a look ahead to look you.
whoah this weblog is great i love reading your articles. Stay up the great work! You realize, a lot of individuals are searching around for this info, you could aid them greatly.
Автор статьи представляет разнообразные факты и статистику, оставляя решение оценки информации читателям. Это сообщение отправлено с сайта https://ru.gototop.ee/
Читателям предоставляется возможность оценить представленные данные и сделать собственные выводы.
I was able to find good information from your blog articles.
Статья помогла мне получить глубокое понимание проблемы, о которой я раньше не задумывался.
Я прочитал эту статью с большим удовольствием! Автор умело смешал факты и личные наблюдения, что придало ей уникальный характер. Я узнал много интересного и наслаждался каждым абзацем. Браво!
Статья хорошо структурирована, что облегчает чтение и понимание.
Автор представляет сложные концепции и понятия в понятной и доступной форме.
Я оцениваю объективность и непредвзятость автора в представлении информации.
Читателям предоставляется возможность самостоятельно исследовать представленные факты и принять собственное мнение.
hello there and thank you for your info – I’ve definitely picked up something new from right here. I did however expertise some technical points using this web site, as I experienced to reload the web site many times previous to I could get it to load properly. I had been wondering if your web hosting is OK? Not that I’m complaining, but slow loading instances times will very frequently affect your placement in google and could damage your high quality score if advertising and marketing with Adwords. Well I am adding this RSS to my email and could look out for a lot more of your respective fascinating content. Make sure you update this again soon.
Автор предоставляет дополнительные ресурсы для тех, кто хочет углубиться в изучение темы.
Статья представляет различные точки зрения и подробно анализирует аргументы каждой стороны.
I am genuinely thankful to the owner of this site who has shared this fantastic article at at this place.
Эта статья просто великолепна! Она представляет информацию в полном объеме и включает в себя практические примеры и рекомендации. Я нашел ее очень полезной и вдохновляющей. Большое спасибо автору за такую выдающуюся работу!
Автор приводит примеры из различных источников, что позволяет получить более полное представление о теме. Статья является нейтральным и информативным ресурсом для тех, кто интересуется данной проблематикой.
Я прочитал эту статью с огромным интересом! Автор умело объединил факты, статистику и персональные истории, что делает ее настоящей находкой. Я получил много новых знаний и вдохновения. Браво!
Я очень доволен, что прочитал эту статью. Она оказалась настоящим открытием для меня. Информация была представлена в увлекательной и понятной форме, и я получил много новых знаний. Спасибо автору за такое удивительное чтение!
For hottest information you have to visit internet and on internet I found this website as a most excellent site for hottest updates.
Great goods from you, man. I have understand your stuff previous to and you are just extremely wonderful. I really like what you have acquired here, really like what you are stating and the way in which you say it. You make it entertaining and you still care for to keep it sensible. I can’t wait to read much more from you. This is actually a tremendous site.
I feel that is among the so much vital info for me. And i’m satisfied reading your article. But want to statement on some general issues, The site taste is wonderful, the articles is in point of fact excellent : D. Excellent job, cheers
Полезно видеть, что статья предоставляет информацию без скрытой агенды или однозначных выводов.
Мне понравилась глубина исследования, проведенного автором, для подкрепления своих утверждений.
I have read so many content on the topic of the blogger lovers but this post is truly a fastidious piece of writing, keep it up.
Статья представляет интересный взгляд на данную тему и содержит ряд полезной информации. Понравилась аккуратная структура и логическое построение аргументов.
Yes! Finally someone writes about keyword1.
Статья содержит практические советы, которые можно применить в реальной жизни.
Статья представляет важные факты и анализирует текущую ситуацию с разных сторон.
Читателям предоставляется возможность обдумать и обсудить представленные факты и аргументы.
Автор статьи предоставляет информацию с разных сторон, представляя факты и аргументы.
Информационная статья предлагает взвешенный подход к обсуждаемой теме, аргументируя свои выводы доказательствами и статистикой.
Читателям предоставляется возможность самостоятельно интерпретировать представленную информацию.
Статья представляет информацию о различных аспектах темы, основываясь на проверенных источниках.
Статья представляет разнообразные аргументы и позиции, основанные на существующих данных и экспертном мнении.
Я оцениваю использование автором разнообразных источников, чтобы подтвердить свои утверждения.
Hi, everything is going fine here and ofcourse every one is sharing facts, that’s actually good, keep up writing.
It’s hard to find well-informed people on this topic, however, you sound like you know what you’re talking about! Thanks
Я хотел бы отметить глубину исследования, представленную в этой статье. Автор не только предоставил факты, но и провел анализ их влияния и последствий. Это действительно ценный и информативный материал!
I was extremely pleased to find this site. I wanted to thank you for ones time just for this wonderful read!! I definitely savored every little bit of it and i also have you bookmarked to check out new stuff in your web site.
That is a really good tip particularly to those fresh to the blogosphere. Brief but very accurate info… Thank you for sharing this one. A must read article!
My partner and I absolutely love your blog and find most of your post’s to be exactly what I’m looking for. Do you offer guest writers to write content available for you? I wouldn’t mind publishing a post or elaborating on many of the subjects you write in relation to here. Again, awesome website!
Мне понравилось объективное представление разных точек зрения на проблему.
Автор использовал разнообразные источники, чтобы подкрепить свои утверждения.
Я просто не могу не поделиться своим восхищением этой статьей! Она является источником ценных знаний, представленных с таким ясным и простым языком. Спасибо автору за его умение сделать сложные вещи доступными!
Статья предлагает объективный обзор исследований, проведенных в данной области. Необходимая информация представлена четко и доступно, что позволяет читателю оценить все аспекты рассматриваемой проблемы.
Hi there to every one, the contents existing at this web page are in fact remarkable for people experience, well, keep up the nice work fellows.
Ahaa, its pleasant conversation regarding this article here at this weblog, I have read all that, so now me also commenting here.
Piece of writing writing is also a excitement, if you know then you can write otherwise it is difficult to write.
Hi there, just became alert to your blog through Google, and found that it’s really informative. I’m going to watch out for brussels. I will appreciate if you continue this in future. Numerous people will be benefited from your writing. Cheers!
What’s up to all, how is all, I think every one is getting more from this web page, and your views are good designed for new people.
Статья представляет важные факты и анализирует текущую ситуацию с разных сторон.
Автор предлагает читателю разные взгляды на проблему, что способствует формированию собственного мнения.
Надеюсь, что эти дополнительные комментарии принесут ещё больше позитивных отзывов на информационную статью! Это сообщение отправлено с сайта GoToTop.ee
Hey There. I discovered your weblog the usage of msn. That is an extremely well written article. I’ll make sure to bookmark it and return to learn extra of your useful information. Thanks for the post. I’ll certainly comeback.
Отличная статья! Я бы хотел отметить ясность и логичность, с которыми автор представил информацию. Это помогло мне легко понять сложные концепции. Большое спасибо за столь прекрасную работу!
Howdy would you mind letting me know which webhost you’re utilizing? I’ve loaded your blog in 3 different web browsers and I must say this blog loads a lot quicker then most. Can you recommend a good hosting provider at a fair price? Many thanks, I appreciate it!
Greetings from California! I’m bored to tears at work so I decided to browse your site on my iphone during lunch break. I enjoy the info you provide here and can’t wait to take a look when I get home. I’m shocked at how quick your blog loaded on my mobile .. I’m not even using WIFI, just 3G .. Anyways, awesome blog!
Надеюсь, что эти комментарии добавят ещё больше позитива и поддержки к информационной статье! Это сообщение отправлено с сайта GoToTop.ee
Конечно, вот ещё несколько положительных комментариев на статью. Это сообщение отправлено с сайта https://ru.gototop.ee/
Мне понравилось разнообразие рассмотренных в статье аспектов проблемы.
Автор предлагает подробное объяснение сложных понятий, связанных с темой.
Автор предлагает рациональные и аргументированные выводы на основе представленных фактов.
Я хотел бы выразить признательность автору этой статьи за его объективный подход к теме. Он представил разные точки зрения и аргументы, что позволило мне получить полное представление о рассматриваемой проблеме. Очень впечатляюще!
I have read so many posts about the blogger lovers but this article is really a good piece of writing, keep it up.
Hello, this weekend is pleasant in favor of me, as this moment i am reading this wonderful informative post here at my residence.
Great work! This is the kind of info that should be shared across the web. Disgrace on Google for no longer positioning this submit higher! Come on over and seek advice from my site . Thank you =)
It is really a great and useful piece of information. I’m glad that you just shared this helpful info with us. Please keep us informed like this. Thank you for sharing.
Автор старается быть объективным и предоставляет достаточно информации для осмысления и дальнейшего обсуждения.
Автор статьи предоставляет информацию в понятной форме, избегая субъективных оценок.
Hi there i am kavin, its my first time to commenting anywhere, when i read this paragraph i thought i could also make comment due to this good paragraph.
I’m gone to convey my little brother, that he should also visit this website on regular basis to get updated from most recent information.
What a data of un-ambiguity and preserveness of valuable know-how regarding unpredicted emotions.
Я благодарен автору этой статьи за его тщательное и глубокое исследование. Он представил информацию с большой детализацией и аргументацией, что делает эту статью надежным источником знаний. Очень впечатляющая работа!
Читателям предоставляется возможность рассмотреть разные аспекты темы и сделать собственные выводы на основе предоставленных данных. Это сообщение отправлено с сайта https://ru.gototop.ee/
Thanks for finally writing about > blog_title < Liked it!
Автор представляет сложные концепции и понятия в понятной и доступной форме.
I do not even understand how I ended up here, but I assumed this submit was once good. I don’t understand who you’re however certainly you’re going to a famous blogger when you aren’t already. Cheers!
WOW just what I was searching for. Came here by searching for meta_keyword
Heya i’m for the first time here. I came across this board and I find It really useful & it helped me out a lot. I hope to give something back and help others like you helped me.
Hi! Quick question that’s totally off topic. Do you know how to make your site mobile friendly? My blog looks weird when browsing from my iphone. I’m trying to find a template or plugin that might be able to resolve this problem. If you have any suggestions, please share. Cheers!
Hi friends, its great post regarding teachingand entirely defined, keep it up all the time.
Автор приводит примеры из различных источников, что позволяет получить более полное представление о теме. Статья является нейтральным и информативным ресурсом для тех, кто интересуется данной проблематикой.
I must thank you for the efforts you’ve put in penning this site. I am hoping to see the same high-grade content by you later on as well. In fact, your creative writing abilities has motivated me to get my very own blog now 😉
Эта статья действительно заслуживает высоких похвал! Она содержит информацию, которую я долго искал, и дает полное представление о рассматриваемой теме. Благодарю автора за его тщательную работу и отличное качество материала!
Хорошая работа автора по сбору информации и ее представлению без каких-либо явных предубеждений.
Читателям предоставляется возможность рассмотреть разные аспекты темы и сделать собственные выводы на основе предоставленных данных. Это сообщение отправлено с сайта https://ru.gototop.ee/
That is a really good tip especially to those new to the blogosphere. Brief but very precise info… Thank you for sharing this one. A must read post!
Я прочитал эту статью с большим удовольствием! Она написана ясно и доступно, несмотря на сложность темы. Большое спасибо автору за то, что делает сложные понятия понятными для всех.
Я оцениваю аккуратность и точность фактов, представленных в статье.
Автор представляет разные точки зрения на проблему и оставляет читателю пространство для собственных размышлений.
Я только что прочитал эту статью, и мне действительно понравилось, как она написана. Автор использовал простой и понятный язык, несмотря на тему, и представил информацию с большой ясностью. Очень вдохновляюще!
Статья содержит дополнительные ресурсы для тех, кто хочет глубже изучить тему.
Автор старается сохранить нейтральность и обеспечить читателей информацией для самостоятельного принятия решений.
Я нашел в статье полезные источники, которые могу изучить для получения дополнительной информации.
Howdy! Would you mind if I share your blog with my myspace group? There’s a lot of folks that I think would really enjoy your content. Please let me know. Thank you
This page certainly has all the information I wanted about this subject and didn’t know who to ask.
Автор статьи предлагает достоверные данные и факты, представленные в нейтральном ключе.
It’s amazing in favor of me to have a site, which is valuable designed for my know-how. thanks admin
Я благодарен автору этой статьи за его тщательное и глубокое исследование. Он представил информацию с большой детализацией и аргументацией, что делает эту статью надежным источником знаний. Очень впечатляющая работа!
Статья содержит информацию, которую можно применить в практической деятельности.
Читателям предоставляется возможность самостоятельно изучить представленные факты и сделать информированный вывод.
Автор предоставляет различные точки зрения и аргументы, что помогает читателю получить полную картину проблемы.
Я хотел бы выразить свою благодарность автору этой статьи за исчерпывающую информацию, которую он предоставил. Я нашел ответы на многие свои вопросы и получил новые знания. Это действительно ценный ресурс!
Автор представляет разнообразные точки зрения на проблему, что помогает читателю получить обширное представление о ней.
Автор предлагает дополнительные ресурсы, которые помогут читателю углубиться в тему и расширить свои знания.
Я хотел бы выразить свою восторженность этой статьей! Она не только информативна, но и вдохновляет меня на дальнейшее изучение темы. Автор сумел передать свою страсть и знания, что делает эту статью поистине уникальной.
Мне понравился нейтральный тон статьи, который позволяет читателю самостоятельно сформировать мнение.
Hello there! I could have sworn I’ve visited this site before but after browsing through some of the posts I realized it’s new to me. Nonetheless, I’m definitely happy I discovered it and I’ll be book-marking it and checking back regularly!
Автор предоставляет анализ последствий проблемы и возможных путей ее решения.
Статья содержит обоснованные аргументы, которые вызывают дальнейшую рефлексию у читателя.
Wow that was unusual. I just wrote an very long comment but after I clicked submit my comment didn’t appear. Grrrr… well I’m not writing all that over again. Regardless, just wanted to say great blog!
At this time I am ready to do my breakfast, afterward having my breakfast coming yet again to read more news.
Надеюсь, что эти комментарии добавят ещё больше позитива и поддержки к информационной статье! Это сообщение отправлено с сайта GoToTop.ee
Статья содержит разнообразные факты и аргументы, представленные в объективной манере.
Автор старается дать читателям достаточно информации для их собственного исследования и принятия решений.
Очень интересная исследовательская работа! Статья содержит актуальные факты, аргументированные доказательствами. Это отличный источник информации для всех, кто хочет поглубже изучить данную тему.
Статья предоставляет полезную информацию, основанную на обширном исследовании.
Magnificent items from you, man. I have have in mind your stuff previous to and you are simply extremely great. I really like what you’ve acquired here, really like what you’re stating and the best way in which you assert it. You’re making it enjoyable and you still care for to keep it sensible. I can not wait to learn far more from you. This is actually a great website.
Статья содержит разнообразные факты и аргументы, представленные в объективной манере.
Fantastic web site. Lots of helpful info here. I’m sending it to several pals ans additionally sharing in delicious. And naturally, thanks on your sweat!
Статья представляет интересный взгляд на данную тему и содержит ряд полезной информации. Понравилась аккуратная структура и логическое построение аргументов.
Статья содержит аргументы, которые помогают читателю лучше понять важность и последствия проблемы.
Everyone loves what you guys are usually up too. This kind of clever work and reporting! Keep up the good works guys I’ve you guys to blogroll.
This design is spectacular! You definitely know how to keep a reader amused. Between your wit and your videos, I was almost moved to start my own blog (well, almost…HaHa!) Great job. I really loved what you had to say, and more than that, how you presented it. Too cool!
Hi, I do believe this is a great blog. I stumbledupon it 😉 I’m going to return yet again since i have saved as a favorite it. Money and freedom is the greatest way to change, may you be rich and continue to guide others.
Автор представил широкий спектр мнений на эту проблему, что позволяет читателям самостоятельно сформировать свое собственное мнение. Полезное чтение для тех, кто интересуется данной темой.
May I simply say what a comfort to find an individual who really knows what they are talking about on the net. You actually realize how to bring an issue to light and make it important. A lot more people must read this and understand this side of the story. I can’t believe you are not more popular since you most certainly have the gift.
Hello to all, because I am in fact eager of reading this webpage’s post to be updated regularly. It consists of nice information.
Hey! I know this is kinda off topic but I’d figured I’d ask. Would you be interested in exchanging links or maybe guest authoring a blog post or vice-versa? My site addresses a lot of the same subjects as yours and I think we could greatly benefit from each other. If you’re interested feel free to shoot me an email. I look forward to hearing from you! Great blog by the way!
Asking questions are really nice thing if you are not understanding something fully, however this paragraph offers nice understanding even.
Wow that was unusual. I just wrote an incredibly long comment but after I clicked submit my comment didn’t show up. Grrrr… well I’m not writing all that over again. Anyway, just wanted to say fantastic blog!
Quality articles is the crucial to interest the users to go to see the site, that’s what this web site is providing.
Это помогает создать обстановку, в которой читатели могут обдумать и обсудить тему без пристрастия.
An outstanding share! I’ve just forwarded this onto a colleague who has been doing a little research on this. And he in fact bought me dinner because I discovered it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanx for spending the time to talk about this subject here on your website.
Автор статьи представляет информацию без предвзятости, предоставляя различные точки зрения и факты.
Hello There. I found your blog using msn. This is a very well written article. I will be sure to bookmark it and return to read more of your useful information. Thanks for the post. I will definitely comeback.
Читателям предоставляется возможность самостоятельно исследовать представленные факты и принять собственное мнение.
Это помогает читателям получить всестороннее представление о теме без явных предубеждений.
Hello to every single one, it’s truly a nice for me to pay a quick visit this web page, it includes helpful Information.
Читателям предоставляется возможность ознакомиться с различными аспектами темы и сделать собственные выводы.
Статья представляет несколько точек зрения на данную тему и анализирует их достоинства и недостатки. Это помогает читателю рассмотреть проблему с разных сторон и принять информированное решение.
Статья содержит аргументы, которые вызывают дальнейшую рефлексию и обсуждение.
Статья содержит ссылки на актуальные и авторитетные источники, что делает ее надежной и достоверной.
Автор предоставляет ссылки на авторитетные источники, что делает статью надежной и достоверной.
Автор представляет свои идеи объективно и не прибегает к эмоциональным уловкам.
If you are going for most excellent contents like myself, just pay a visit this web page all the time since it gives quality contents, thanks
Very nice post. I just stumbled upon your weblog and wished to say that I’ve truly enjoyed surfing around your blog posts. After all I’ll be subscribing on your feed and I’m hoping you write again very soon!
Статья представляет широкий обзор темы, учитывая различные аспекты проблемы.
Я чувствую, что эта статья является настоящим источником вдохновения. Она предлагает новые идеи и вызывает желание узнать больше. Большое спасибо автору за его творческий и информативный подход!
Статья представляет интересный взгляд на данную тему и содержит ряд полезной информации. Понравилась аккуратная структура и логическое построение аргументов.
Автор предоставляет актуальную информацию, которая помогает читателю быть в курсе последних событий и тенденций.
Thankfulness to my father who told me regarding this blog, this website is actually amazing.
Я нашел в статье полезные источники, которые могу изучить для получения дополнительной информации.
Я оцениваю объективность и сбалансированность подхода автора к представлению информации.
Статья представляет все ключевые аспекты темы, обеспечивая при этом достаточную детализацию.
Статья помогла мне лучше понять контекст и значение проблемы в современном обществе.
Это поддерживается ссылками на надежные источники, что делает статью достоверной и нейтральной.
Thanks for the good writeup. It if truth be told was a amusement account it. Look complex to more introduced agreeable from you! By the way, how could we communicate?
I blog often and I really appreciate your information. This article has truly peaked my interest. I’m going to bookmark your blog and keep checking for new information about once a week. I subscribed to your RSS feed too.
Это позволяет читателям самостоятельно оценить и проанализировать информацию.
Автор старается сохранить нейтральность, чтобы читатели могли основываться на объективной информации при формировании своего мнения. Это сообщение отправлено с сайта https://ru.gototop.ee/
Hey There. I found your blog using msn. This is a very well written article. I’ll be sure to bookmark it and return to read more of your useful info. Thanks for the post. I’ll definitely comeback.
It’s actually a great and useful piece of information. I am glad that you just shared this useful info with us. Please stay us up to date like this. Thank you for sharing.
Saved as a favorite, I like your blog!
Статья содержит обширный объем информации, которая подкреплена соответствующими доказательствами.
Статья содержит аргументы, которые помогают читателю лучше понять важность и последствия проблемы.
Статья помогла мне получить новые знания и пересмотреть свое представление о проблеме.
Do you mind if I quote a couple of your posts as long as I provide credit and sources back to your blog? My website is in the very same niche as yours and my users would definitely benefit from a lot of the information you present here. Please let me know if this ok with you. Thanks a lot!
Я оцениваю объективный подход автора и его стремление представить полную картину проблемы.
Статья предлагает широкий обзор темы, представляя разные точки зрения и подробности.
Эта статья действительно заслуживает высоких похвал! Она содержит информацию, которую я долго искал, и дает полное представление о рассматриваемой теме. Благодарю автора за его тщательную работу и отличное качество материала!
Hi, i read your blog from time to time and i own a similar one and i was just curious if you get a lot of spam responses? If so how do you protect against it, any plugin or anything you can suggest? I get so much lately it’s driving me crazy so any help is very much appreciated.
Я прочитал эту статью с большим удовольствием! Она написана ясно и доступно, несмотря на сложность темы. Большое спасибо автору за то, что делает сложные понятия понятными для всех.
Автор предлагает анализ преимуществ и недостатков разных подходов к решению проблемы.
Я оцениваю объективность и непредвзятость автора в представлении информации.
Я ценю балансировку автора в описании проблемы. Он предлагает читателю достаточно аргументов и контекста для формирования собственного мнения, не внушая определенную точку зрения.
I’ve read several good stuff here. Certainly price bookmarking for revisiting. I surprise how much attempt you put to make this type of fantastic informative website.
That is a great tip particularly to those new to the blogosphere. Brief but very accurate info… Appreciate your sharing this one. A must read article!
Конечно, вот ещё несколько положительных комментариев на статью. Это сообщение отправлено с сайта https://ru.gototop.ee/
Статья содержит актуальную информацию, которая помогает понять сложность и важность проблемы.
Автор статьи представляет информацию с акцентом на факты и статистику, не высказывая предпочтений.
Очень интересная исследовательская работа! Статья содержит актуальные факты, аргументированные доказательствами. Это отличный источник информации для всех, кто хочет поглубже изучить данную тему.
Howdy just wanted to give you a brief heads up and let you know a few of the images aren’t loading properly. I’m not sure why but I think its a linking issue. I’ve tried it in two different internet browsers and both show the same outcome.
Автор предлагает читателю дополнительные ресурсы для глубокого погружения в тему.
Аргументы подкреплены фактами и исследованиями, что позволяет читателям рассмотреть разные стороны вопроса.
Полезно, что автор предоставил разные точки зрения на обсуждаемую тему.
Автор представляет информацию в легком и доступном формате, что делает ее приятной для чтения.
Автор статьи предоставляет подробные факты и данные, не выражая собственного мнения.
Статья представляет несколько точек зрения на данную тему и анализирует их достоинства и недостатки. Это помогает читателю рассмотреть проблему с разных сторон и принять информированное решение.
Надеюсь, что эти комментарии добавят ещё больше позитива и поддержки к информационной статье!
Надеюсь, вам понравятся и эти комментарии!
Автор предоставляет анализ достоинств и недостатков различных подходов к решению проблемы.
Надеюс
I’ll right away seize your rss feed as I can’t find your email subscription link or e-newsletter service. Do you have any? Please allow me recognise in order that I may just subscribe. Thanks.
Я благодарен автору этой статьи за его тщательное и глубокое исследование. Он представил информацию с большой детализацией и аргументацией, что делает эту статью надежным источником знаний. Очень впечатляющая работа!
Appreciation to my father who shared with me regarding this blog, this blog is genuinely awesome.
bookmarked!!, I like your site!
Автор статьи представляет информацию, основанную на разных источниках и экспертных мнениях.
I just like the helpful information you provide on your articles. I’ll bookmark your blog and take a look at once more right here regularly. I am slightly sure I will be informed plenty of new stuff right here! Good luck for the following!
May I just say what a comfort to uncover someone that really knows what they’re discussing on the web. You definitely know how to bring a problem to light and make it important. More and more people have to check this out and understand this side of the story. It’s surprising you are not more popular since you most certainly have the gift.
Статья помогла мне лучше понять контекст и значение проблемы в современном обществе.
Wonderful items from you, man. I’ve be mindful your stuff previous to and you are simply too excellent. I really like what you have obtained here, certainly like what you are stating and the way in which through which you say it. You’re making it entertaining and you still care for to stay it sensible. I cant wait to read far more from you. This is really a terrific site.
Thank you for another magnificent article. Where else could anybody get that type of info in such an ideal method of writing? I have a presentation subsequent week, and I’m at the look for such information.
Статья предлагает конкретные примеры, чтобы проиллюстрировать свои аргументы.
Link exchange is nothing else except it is only placing the other person’s weblog link on your page at suitable place and other person will also do same in support of you.
Автор представляет сложные концепции и понятия в понятной и доступной форме.
Hi, i read your blog from time to time and i own a similar one and i was just wondering if you get a lot of spam responses? If so how do you stop it, any plugin or anything you can suggest? I get so much lately it’s driving me crazy so any support is very much appreciated.
Статья содержит информацию, основанную на достоверных источниках и экспертных мнениях.
I think the admin of this web site is actually working hard in favor of his web page, since here every material is quality based stuff.
Автор представляет сложные темы в понятной и доступной форме для широкой аудитории.
Автор статьи предоставляет факты и аргументы, не влияя на читателя своими собственными предпочтениями или предвзятостью.
I want to to thank you for this wonderful read!! I certainly loved every little bit of it. I’ve got you book-marked to look at new things you post…
Whoa! This blog looks exactly like my old one! It’s on a entirely different topic but it has pretty much the same layout and design. Wonderful choice of colors!
Статья представляет различные точки зрения и подробно анализирует аргументы каждой стороны.
Я оцениваю степень детализации информации в статье, которая позволяет получить полное представление о проблеме.
Автор старается представить материал нейтрально, оставляя пространство для собственного рассмотрения и анализа.
Я оцениваю тщательность и точность, с которыми автор подошел к составлению этой статьи. Он привел надежные источники и представил информацию без преувеличений. Благодаря этому, я могу доверять ей как надежному источнику знаний.
Hi there, this weekend is nice in support of me, since this occasion i am reading this impressive informative piece of writing here at my house.
Это помогает читателям получить полное представление о сложности и многогранности обсуждаемой темы.
If you are going for finest contents like me, only pay a visit this web site all the time since it offers quality contents, thanks
Автор статьи хорошо структурировал информацию и представил ее в понятной форме.
Статья представляет анализ разных точек зрения на проблему, что помогает читателю получить полное представление о ней.
Мне понравился баланс между фактами и мнениями в статье.
Hi, i feel that i saw you visited my blog thus i got here to return the desire?.I am attempting to find things to improve my website!I assume its ok to make use of some of your ideas!!
Hello! I could have sworn I’ve visited this web site before but after going through a few of the posts I realized it’s new to me. Anyhow, I’m definitely delighted I discovered it and I’ll be book-marking it and checking back regularly!
Я нашел эту статью чрезвычайно познавательной и вдохновляющей. Автор обладает уникальной способностью объединять различные идеи и концепции, что делает его работу по-настоящему ценной и полезной.
Я не могу не отметить стиль и ясность изложения в этой статье. Автор использовал простой и понятный язык, что помогло мне легко усвоить материал. Огромное спасибо за такой доступный подход!
Автор представил широкий спектр мнений на эту проблему, что позволяет читателям самостоятельно сформировать свое собственное мнение. Полезное чтение для тех, кто интересуется данной темой.
Читателям предоставляется возможность самостоятельно интерпретировать представленную информацию.
Good info. Lucky me I recently found your website by accident (stumbleupon). I’ve saved as a favorite for later!
Статья предлагает конкретные примеры, чтобы проиллюстрировать свои аргументы.
Я хотел бы выразить признательность автору за его глубокое понимание темы и его способность представить информацию во всей ее полноте. Я по-настоящему насладился этой статьей и узнал много нового!
Читателям предоставляется возможность обдумать и обсудить представленные факты и аргументы.
Я оцениваю тщательность и точность исследования, представленного в этой статье. Автор провел глубокий анализ и представил аргументированные выводы. Очень важная и полезная работа!
you’re in point of fact a good webmaster. The site loading speed is incredible. It kind of feels that you’re doing any unique trick. Furthermore, The contents are masterwork. you have done a magnificent activity in this subject!
Автор статьи представляет разнообразные факты и статистику, оставляя решение оценки информации читателям. Это сообщение отправлено с сайта https://ru.gototop.ee/
Автор представил широкий спектр мнений на эту проблему, что позволяет читателям самостоятельно сформировать свое собственное мнение. Полезное чтение для тех, кто интересуется данной темой.
Great delivery. Sound arguments. Keep up the great work.
Хорошая статья, в которой автор предлагает различные точки зрения и аргументы.
I could not resist commenting. Well written!
You’ve made some decent points there. I looked on the net for additional information about the issue and found most individuals will go along with your views on this website.
Я бы хотел выразить свою благодарность автору этой статьи за его профессионализм и преданность точности. Он предоставил достоверные факты и аргументированные выводы, что делает эту статью надежным источником информации.
Он/она не стремится принимать сторону и предоставляет читателям возможность самостоятельно сделать выводы.
Эта статья оказалась исключительно информативной и понятной. Автор представил сложные концепции и теории в простой и доступной форме. Я нашел ее очень полезной и вдохновляющей!
Автор старается сохранить нейтральность и оставляет решение оценки информации читателям.
Автор старается сохранить нейтральность, предоставляя обстоятельную основу для дальнейшего рассмотрения темы.
Это позволяет читателям самостоятельно сформировать свое мнение.
Я восхищен тем, как автор умело объясняет сложные концепции. Он сумел сделать информацию доступной и интересной для широкой аудитории. Это действительно заслуживает похвалы!
Я не могу не отметить стиль и ясность изложения в этой статье. Автор использовал простой и понятный язык, что помогло мне легко усвоить материал. Огромное спасибо за такой доступный подход!
I do not even know the way I stopped up here, however I assumed this publish was great. I do not know who you might be but certainly you’re going to a well-known blogger when you aren’t already. Cheers!
Genuinely when someone doesn’t understand then its up to other visitors that they will assist, so here it happens.
Эта статья превзошла мои ожидания! Она содержит обширную информацию, иллюстрирует примерами и предлагает практические советы. Я благодарен автору за его усилия в создании такого полезного материала.
Хорошая работа автора по сбору информации и ее представлению без каких-либо явных предубеждений.
Я оцениваю четкую структуру статьи, которая помогает организовать мысли и понять ее содержание.
I just couldn’t go away your web site prior to suggesting that I really enjoyed the usual info an individual provide for your visitors? Is going to be again incessantly to investigate cross-check new posts
What’s up friends, how is everything, and what you wish for to say regarding this post, in my view its in fact amazing in favor of me.
Keep this going please, great job!
Hello, I enjoy reading through your article post. I like to write a little comment to support you.
Hey there! I know this is somewhat off topic but I was wondering which blog platform are you using for this site? I’m getting fed up of WordPress because I’ve had issues with hackers and I’m looking at alternatives for another platform. I would be awesome if you could point me in the direction of a good platform.
Я прочитал эту статью с большим удовольствием! Автор умело смешал факты и личные наблюдения, что придало ей уникальный характер. Я узнал много интересного и наслаждался каждым абзацем. Браво!
Я просто не могу пройти мимо этой статьи без оставления положительного комментария. Она является настоящим примером качественной журналистики и глубокого исследования. Очень впечатляюще!
Очень хорошо организованная статья! Автор умело структурировал информацию, что помогло мне легко следовать за ней. Я ценю его усилия в создании такого четкого и информативного материала.
I have been surfing online more than 3 hours today, yet I never found any interesting article like yours. It’s pretty worth enough for me. In my opinion, if all website owners and bloggers made good content as you did, the internet will be much more useful than ever before.
Автор представляет сложные понятия в понятной и доступной форме, что erleichtert das Verständnis.
Ridiculous story there. What occurred after? Take care!
Hi there mates, how is all, and what you desire to say regarding this article, in my view its in fact remarkable designed for me.
Автор старается быть нейтральным, предоставляя читателям возможность самих оценить представленные доводы.
Автор предоставляет разнообразные источники, которые дополняют и расширяют представленную информацию.
Статья помогла мне лучше понять сложные аспекты темы, с которыми я ранее не сталкивался.
Статья помогла мне расширить свои знания и глубже понять рассматриваемую тему.
After looking at a handful of the blog posts on your site, I honestly like your way of blogging. I bookmarked it to my bookmark webpage list and will be checking back soon. Take a look at my web site too and let me know what you think.
Эта статья – источник ценной информации! Я оцениваю глубину исследования и разнообразие рассматриваемых аспектов. Она действительно расширила мои знания и помогла мне лучше понять тему. Большое спасибо автору за такую качественную работу!
Статья предлагает глубокий анализ темы и рассматривает ее со всех сторон.
It’s in fact very complex in this full of activity life to listen news on TV, so I simply use web for that purpose, and obtain the latest information.
Я восхищен тем, как автор умело объясняет сложные концепции. Он сумел сделать информацию доступной и интересной для широкой аудитории. Это действительно заслуживает похвалы!
Я впечатлен этой статьей! Она не только информативна, но и вдохновляющая. Мне понравился подход автора к обсуждению темы, и я узнал много нового. Огромное спасибо за такую интересную и полезную статью!
Статья представляет интересный взгляд на данную тему и содержит ряд полезной информации. Понравилась аккуратная структура и логическое построение аргументов.
Я оцениваю тщательность и точность исследования, представленного в этой статье. Автор провел глубокий анализ и представил аргументированные выводы. Очень важная и полезная работа!
What’s up to all, because I am really eager of reading this weblog’s post to be updated regularly. It includes fastidious information.
Статья представляет аккуратный обзор современных исследований и различных точек зрения на данную проблему. Она предоставляет хороший стартовый пункт для тех, кто хочет изучить тему более подробно.
Hey there outstanding blog! Does running a blog similar to this take a large amount of work? I’ve virtually no expertise in programming but I had been hoping to start my own blog in the near future. Anyhow, should you have any recommendations or tips for new blog owners please share. I know this is off subject however I simply needed to ask. Thanks a lot!
Это помогает читателям получить полное представление о сложности и многообразии данного вопроса.
Читателям предоставляется возможность ознакомиться с различными аспектами и сделать собственные выводы.
Amazing! Its genuinely awesome article, I have got much clear idea on the topic of from this post.
I don’t know if it’s just me or if everybody else encountering problems with your blog. It appears as if some of the text within your content are running off the screen. Can somebody else please provide feedback and let me know if this is happening to them too? This could be a problem with my web browser because I’ve had this happen before. Kudos
Я оцениваю тщательность и качество исследования, представленного в этой статье. Автор предоставил надежные источники и учел различные аспекты темы. Это действительно ценный ресурс для всех интересующихся.
Мне понравилось разнообразие рассмотренных в статье аспектов проблемы.
Эта статья является настоящим сокровищем информации. Я был приятно удивлен ее глубиной и разнообразием подходов к рассматриваемой теме. Спасибо автору за такой тщательный анализ и интересные факты!
Мне понравился нейтральный тон статьи, который позволяет читателю самостоятельно сформировать мнение.
Статья содержит актуальную информацию, которая помогает разобраться в современных тенденциях и проблемах.
Автор использует ясные и доступные примеры, чтобы проиллюстрировать свои аргументы.
Автор предлагает аргументы, подкрепленные проверенными источниками.
Wow, this paragraph is fastidious, my younger sister is analyzing these kinds of things, so I am going to inform her.
Автор статьи представляет информацию, основанную на достоверных источниках.
Я ценю информативный подход этой статьи. Она предоставляет достаточно фактов и данных для лучшего понимания проблемы. Хотелось бы увидеть больше ссылок на исследования и источники информации.
It’s in fact very complicated in this active life to listen news on TV, therefore I just use world wide web for that purpose, and take the most up-to-date information.
You actually make it seem so easy with your presentation but I find this topic to be actually something that I think I would never understand. It seems too complicated and very broad for me. I’m looking forward for your next post, I’ll try to get the hang of it!
I blog frequently and I genuinely appreciate your content. Your article has really peaked my interest. I’m going to book mark your website and keep checking for new details about once per week. I subscribed to your Feed too.
Мне понравилась объективность автора и его способность представить информацию без предвзятости.
Статья содержит анализ причин и последствий проблемы, что позволяет лучше понять ее важность и сложность.
you are in reality a good webmaster. The site loading speed is amazing. It seems that you are doing any unique trick. In addition, The contents are masterwork. you have performed a wonderful process in this matter!
Статья представляет разнообразные точки зрения на обсуждаемую тему и не принимает сторону.
Hi, this weekend is good in support of me, as this moment i am reading this enormous informative post here at my home.
Автор старается подойти к теме нейтрально, чтобы предоставить читателям полную картину.
Excellent blog here! Also your web site loads up very fast! What web host are you using? Can I get your affiliate link to your host? I wish my site loaded up as quickly as yours lol
I could not resist commenting. Very well written!
I’ll right away take hold of your rss as I can not to find your email subscription link or newsletter service. Do you have any? Please permit me recognise in order that I may just subscribe. Thanks.
Автор представляет сложные понятия в понятной и доступной форме, что erleichtert das Verständnis.
Я восхищен тем, как автор умело объясняет сложные концепции. Он сумел сделать информацию доступной и интересной для широкой аудитории. Это действительно заслуживает похвалы!
Автор представил широкий спектр мнений на эту проблему, что позволяет читателям самостоятельно сформировать свое собственное мнение. Полезное чтение для тех, кто интересуется данной темой.
What’s Happening i’m new to this, I stumbled upon this I’ve discovered It absolutely helpful and it has aided me out loads. I’m hoping to give a contribution & help other users like its helped me. Good job.
Автор старается сохранить нейтральность и оставляет решение оценки информации читателям.
Nice post. I used to be checking continuously this blog and I’m inspired! Extremely useful information particularly the last section 🙂 I handle such info much. I used to be looking for this particular info for a long time. Thank you and best of luck.
I got this website from my friend who shared with me on the topic of this web site and at the moment this time I am visiting this web site and reading very informative content at this place.
Информационная статья предлагает всесторонний обзор ситуации, с учетом разных аспектов и аргументов.
Wow, this paragraph is good, my younger sister is analyzing such things, so I am going to let know her.
Статья представляет широкий спектр точек зрения на проблему, что способствует более глубокому пониманию.
Hmm is anyone else having problems with the pictures on this blog loading? I’m trying to figure out if its a problem on my end or if it’s the blog. Any feedback would be greatly appreciated.
Я оцениваю умение автора использовать разнообразные источники, чтобы подкрепить свои утверждения.
Автор статьи представляет информацию без предвзятости, предоставляя различные точки зрения и факты.
Я ценю балансировку автора в описании проблемы. Он предлагает читателю достаточно аргументов и контекста для формирования собственного мнения, не внушая определенную точку зрения.
Quality content is the important to attract the users to pay a visit the site, that’s what this website is providing.
Это позволяет читателям формировать свою собственную точку зрения на основе фактов.
Thanks for another informative website. Where else could I am getting that type of information written in such an ideal manner? I’ve a undertaking that I am just now operating on, and I have been at the glance out for such information.
Hello, for all time i used to check website posts here early in the break of day, since i like to learn more and more.
Это помогает читателям получить объективное представление о рассматриваемой теме.
I’m gone to say to my little brother, that he should also go to see this weblog on regular basis to take updated from latest information.
Hey there would you mind letting me know which webhost you’re using? I’ve loaded your blog in 3 different internet browsers and I must say this blog loads a lot quicker then most. Can you recommend a good hosting provider at a fair price? Cheers, I appreciate it!
Wow, superb blog layout! How long have you been blogging for? you make blogging look easy. The overall look of your web site is magnificent, let alone the content!
Это помогает читателям получить объективное представление о рассматриваемой теме.
Do you mind if I quote a couple of your posts as long as I provide credit and sources back to your site? My blog site is in the exact same area of interest as yours and my users would really benefit from a lot of the information you provide here. Please let me know if this okay with you. Thank you!
Автор статьи представляет разнообразные факты и статистику, оставляя решение оценки информации читателям. Это сообщение отправлено с сайта https://ru.gototop.ee/
Thanks for your marvelous posting! I actually enjoyed reading it, you can be a great author. I will be sure to bookmark your blog and definitely will come back sometime soon. I want to encourage yourself to continue your great work, have a nice evening!
I love your blog.. very nice colors & theme. Did you make this website yourself or did you hire someone to do it for you? Plz respond as I’m looking to create my own blog and would like to find out where u got this from. thank you
Я хотел бы выразить свою благодарность автору этой статьи за исчерпывающую информацию, которую он предоставил. Я нашел ответы на многие свои вопросы и получил новые знания. Это действительно ценный ресурс!
Автор старается быть нейтральным, предоставляя читателям возможность самих оценить представленные доводы.
I seriously love your blog.. Excellent colors & theme. Did you develop this amazing site yourself? Please reply back as I’m planning to create my very own blog and want to find out where you got this from or exactly what the theme is called. Many thanks!
Я хотел бы выразить свою благодарность автору этой статьи за исчерпывающую информацию, которую он предоставил. Я нашел ответы на многие свои вопросы и получил новые знания. Это действительно ценный ресурс!
Attractive section of content. I simply stumbled upon your blog and in accession capital to assert that I get in fact loved account your blog posts. Any way I will be subscribing to your feeds or even I achievement you access persistently rapidly.
Thanks very nice blog!
My spouse and I stumbled over here from a different web page and thought I might check things out. I like what I see so now i’m following you. Look forward to going over your web page yet again.
Автор статьи представляет информацию, основанную на разных источниках и экспертных мнениях.
What’s up to all, since I am in fact eager of reading this web site’s post to be updated regularly. It contains nice data.
Я хотел бы выразить свою благодарность автору за его глубокие исследования и ясное изложение. Он сумел объединить сложные концепции и представить их в доступной форме. Это действительно ценный ресурс для всех, кто интересуется этой темой.
Я оцениваю тщательность и качество исследования, представленного в этой статье. Автор предоставил надежные источники и учел различные аспекты темы. Это действительно ценный ресурс для всех интересующихся.
Статья представляет различные аспекты темы и помогает получить полную картину.
Heya i am for the first time here. I found this board and I find It really useful & it helped me out a lot. I hope to give something back and aid others like you aided me.
I have fun with, result in I discovered just what I was looking for. You have ended my four day long hunt! God Bless you man. Have a great day. Bye
Статья содержит полезную информацию, которая может быть полезной для практического применения.
Я оцениваю четкость и последовательность изложения информации в статье.
Эта статья – источник вдохновения и новых знаний! Я оцениваю уникальный подход автора и его способность представить информацию в увлекательной форме. Это действительно захватывающее чтение!
Автор предлагает дополнительные ресурсы, которые помогут читателю углубиться в тему и расширить свои знания.
Эта статья превзошла мои ожидания! Она содержит обширную информацию, иллюстрирует примерами и предлагает практические советы. Я благодарен автору за его усилия в создании такого полезного материала.
Эта статья – источник вдохновения и новых знаний! Я оцениваю уникальный подход автора и его способность представить информацию в увлекательной форме. Это действительно захватывающее чтение!
Thanks for sharing such a pleasant opinion, post is good, thats why i have read it fully
Автор представляет сложные темы в понятной и доступной форме для широкой аудитории.
What’s Taking place i’m new to this, I stumbled upon this I have discovered It absolutely useful and it has helped me out loads. I am hoping to contribute & aid other users like its helped me. Good job.
I think everything posted made a ton of sense. However, think about this, what if you composed a catchier title? I ain’t saying your information is not good, but suppose you added something that makes people want more? I mean BLOG_TITLE is kinda vanilla. You should glance at Yahoo’s home page and watch how they create post titles to get viewers to click. You might add a related video or a related pic or two to grab readers interested about everything’ve got to say. Just my opinion, it might make your posts a little livelier.
Статья содержит ясные и убедительные аргументы, подкрепленные конкретными примерами.
Я бы хотел отметить актуальность и релевантность этой статьи. Автор предоставил нам свежую и интересную информацию, которая помогает понять современные тенденции и развитие в данной области. Большое спасибо за такой информативный материал!
Это позволяет читателям самостоятельно оценить представленную информацию и сделать информированные выводы.
Информационная статья предлагает всесторонний обзор ситуации, с учетом разных аспектов и аргументов.
Hello to every body, it’s my first go to see of this webpage; this blog contains awesome and truly excellent data in support of readers.
Hi to every body, it’s my first visit of this website; this webpage carries awesome and really fine material in favor of visitors.
Way cool! Some very valid points! I appreciate you writing this post and also the rest of the site is also really good.
Автор предлагает аргументы, подкрепленные проверенными фактами и авторитетными источниками.
excellent issues altogether, you just gained a new reader. What would you recommend about your publish that you made some days ago? Any positive?
Статья содержит дополнительные примеры, которые помогают проиллюстрировать основные концепции.
Он/она не стремится принимать сторону и предоставляет читателям возможность самостоятельно сделать выводы.
Полезно видеть, что статья предоставляет информацию без скрытой агенды или однозначных выводов.
There’s certainly a lot to find out about this topic. I like all of the points you made.
Отлично, что автор обратил внимание на разные точки зрения и представил их в сбалансированном виде.
Статья предлагает широкий обзор событий и фактов, связанных с обсуждаемой темой.
Это помогает читателям осознать сложность проблемы и самостоятельно сформировать свое собственное мнение.
Очень интересная статья! Я был поражен ее актуальностью и глубиной исследования. Автор сумел объединить различные точки зрения и представить полную картину темы. Браво за такой информативный материал!
Я прочитал эту статью с огромным интересом! Автор умело объединил факты, статистику и персональные истории, что делает ее настоящей находкой. Я получил много новых знаний и вдохновения. Браво!
Статья содержит интересные факты, которые помогают глубже понять тему.
Я очень доволен, что прочитал эту статью. Она оказалась настоящим открытием для меня. Информация была представлена в увлекательной и понятной форме, и я получил много новых знаний. Спасибо автору за такое удивительное чтение!
Статья помогла мне лучше понять сложную тему.
Это способствует более глубокому пониманию темы и формированию информированного мнения.
Читатели имеют возможность самостоятельно проанализировать представленные факты и сделать собственные выводы.
Я оцениваю объективность автора и его стремление представить разные точки зрения на проблему.
Great site you have got here.. It’s hard to find high quality writing like yours nowadays. I seriously appreciate individuals like you! Take care!!
This text is worth everyone’s attention. Where can I find out more?
Статья предоставляет информацию из разных источников, обеспечивая балансированное представление фактов и аргументов.
Автор не высказывает собственных предпочтений, что позволяет читателям самостоятельно сформировать свое мнение.
Автор старается представить информацию объективно и позволяет читателям самостоятельно сделать выводы.
We are a group of volunteers and starting a new scheme in our community. Your website provided us with valuable information to work on. You have done a formidable job and our entire community will be thankful to you.
Увеличение ссылочной массы: Ключевой фактор в росте DR сайта. Увеличение ссылочной массы является неотъемлемой частью стратегии роста DR сайта. Этот процесс требует систематического и длительного подхода, включающего в себя поиск высококачественных ссылок, мониторинг и анализ ссылочной массы, а также адаптацию к постоянно меняющемуся окружению поисковых систем. Понимание этих аспектов и их правильная реализация помогут сайту достичь более высокого DR, что в свою очередь улучшит его ранжирование, привлечет больше органического трафика и повысит успех онлайн-присутствия бизнеса.
Мне понравилась систематическая структура статьи, которая позволяет читателю легко следовать логике изложения.
Remarkable issues here. I am very satisfied to look your post. Thank you so much and I’m looking ahead to contact you. Will you kindly drop me a mail?
I’m curious to find out what blog platform you are working with? I’m experiencing some minor security problems with my latest blog and I would like to find something more risk-free. Do you have any solutions?
Статья представляет информацию о текущих событиях, описывая различные аспекты ситуации.
Статья помогает читателю разобраться в сложной проблеме, предлагая разные подходы к ее решению.
Your method of telling all in this paragraph is actually fastidious, all be able to easily know it, Thanks a lot.
Hi there! I know this is kind of off topic but I was wondering if you knew where I could locate a captcha plugin for my comment form? I’m using the same blog platform as yours and I’m having problems finding one? Thanks a lot!
Hey there would you mind sharing which blog platform you’re working with? I’m going to start my own blog in the near future but I’m having a tough time deciding between BlogEngine/Wordpress/B2evolution and Drupal. The reason I ask is because your layout seems different then most blogs and I’m looking for something completely unique. P.S Apologies for being off-topic but I had to ask!
Статья представляет информацию о различных аспектах темы, основываясь на проверенных источниках.
Это поддерживается ссылками на надежные источники, что делает статью достоверной и нейтральной.
Hello there, I found your blog via Google even as looking for a related matter, your site came up, it seems good. I’ve bookmarked it in my google bookmarks.
Надеюсь, что эти комментарии добавят ещё больше положительных настроений к информационной статье!
Я впечатлен этой статьей! Она не только информативна, но и вдохновляющая. Мне понравился подход автора к обсуждению темы, и я узнал много нового. Огромное спасибо за такую интересную и полезную статью!
Очень хорошо исследованная статья! Она содержит много подробностей и является надежным источником информации. Я оцениваю автора за его тщательную работу и приветствую его старания в предоставлении читателям качественного контента.
Мне понравилась четкая структура статьи, которая помогает легко ориентироваться в тексте.
Автор старается представить материал нейтрально, что способствует обоснованному рассмотрению темы.
Heya i am for the first time here. I found this board and I find It really useful & it helped me out a lot. I hope to give something back and help others like you helped me.
Статья является исчерпывающим и объективным рассмотрением темы.
Greetings! Very helpful advice in this particular article! It is the little changes which will make the greatest changes. Thanks a lot for sharing!
Автор хорошо подготовился к теме и представил разнообразные факты.
Hey There. I found your weblog using msn. This is a very smartly written article. I’ll make sure to bookmark it and return to learn extra of your useful info. Thanks for the post. I will certainly return.
Статья предлагает обширный обзор темы, представляя разные точки зрения и аргументы.
Мне понравилась четкая и логическая структура статьи, которая облегчает чтение.
Статья содержит анализ причин возникновения проблемы и возможных путей ее решения.
Я восхищен тем, как автор умело объясняет сложные концепции. Он сумел сделать информацию доступной и интересной для широкой аудитории. Это действительно заслуживает похвалы!
Статья предоставляет информацию из разных источников, обеспечивая балансированное представление фактов и аргументов.
Автор старается представить информацию объективно и позволяет читателям самостоятельно сделать выводы.
Эта статья оказалась исключительно информативной и понятной. Автор представил сложные концепции и теории в простой и доступной форме. Я нашел ее очень полезной и вдохновляющей!
Howdy just wanted to give you a quick heads up and let you know a few of the pictures aren’t loading properly. I’m not sure why but I think its a linking issue. I’ve tried it in two different internet browsers and both show the same results.
Nice post. I learn something totally new and challenging on blogs I stumbleupon everyday. It will always be interesting to read through articles from other authors and practice a little something from their websites.
Эта статья просто великолепна! Она представляет информацию в полном объеме и включает в себя практические примеры и рекомендации. Я нашел ее очень полезной и вдохновляющей. Большое спасибо автору за такую выдающуюся работу!
Статья представляет аккуратный обзор современных исследований и различных точек зрения на данную проблему. Она предоставляет хороший стартовый пункт для тех, кто хочет изучить тему более подробно.
Hi! This post could not be written any better! Reading this post reminds me of my previous room mate! He always kept talking about this. I will forward this post to him. Pretty sure he will have a good read. Many thanks for sharing!
Я восхищен тем, как автор умело объясняет сложные концепции. Он сумел сделать информацию доступной и интересной для широкой аудитории. Это действительно заслуживает похвалы!
Hey! Do you know if they make any plugins to protect against hackers? I’m kinda paranoid about losing everything I’ve worked hard on. Any tips?
Hey There. I found your blog using msn. This is a really well written article. I will make sure to bookmark it and return to read more of your useful info. Thanks for the post. I’ll definitely comeback.
Это позволяет читателям самостоятельно сделать выводы и продолжить исследование по данному вопросу.
For most recent news you have to pay a visit internet and on web I found this website as a finest web page for latest updates.
I was recommended this web site via my cousin. I am no longer sure whether this post is written by him as no one else realize such distinct about my difficulty. You’re wonderful! Thank you!
Wonderful goods from you, man. I have understand your stuff previous to and you’re just too magnificent. I really like what you’ve acquired here, certainly like what you’re saying and the way in which you say it. You make it enjoyable and you still take care of to keep it smart. I cant wait to read much more from you. This is really a great website.
Я ценю балансировку автора в описании проблемы. Он предлагает читателю достаточно аргументов и контекста для формирования собственного мнения, не внушая определенную точку зрения.
Автор статьи предоставляет информацию с разных сторон, представляя факты и аргументы.
Paragraph writing is also a fun, if you be acquainted with after that you can write or else it is complex to write.
Hello There. I found your blog using msn. This is a very well written article. I will be sure to bookmark it and come back to read more of your useful information. Thanks for the post. I will definitely comeback.
Очень хорошо структурированная статья! Я оцениваю ясность и последовательность изложения. Благодаря этому, я смог легко следовать за логикой и усвоить представленную информацию. Большое спасибо автору за такой удобный формат!
Статья предлагает читателям широкий спектр информации, основанной на разных источниках.
Good day! Would you mind if I share your blog with my myspace group? There’s a lot of people that I think would really appreciate your content. Please let me know. Cheers
Nice post. I learn something totally new and challenging on blogs I stumbleupon every day. It will always be helpful to read through content from other writers and practice a little something from their websites.
Я благодарен автору этой статьи за его тщательное и глубокое исследование. Он представил информацию с большой детализацией и аргументацией, что делает эту статью надежным источником знаний. Очень впечатляющая работа!
Статья предлагает разнообразные подходы к решению проблемы и позволяет читателю выбрать наиболее подходящий для него.
Right away I am going away to do my breakfast, once having my breakfast coming yet again to read more news.
Я не могу не отметить качество исследования, представленного в этой статье. Она обогатила мои знания и вдохновила меня на дальнейшее изучение темы. Благодарю автора за его ценный вклад!
Статья представляет факты и аналитические данные, не выражая предпочтений.
Hi, just wanted to tell you, I liked this post. It was practical. Keep on posting!
I love it when people come together and share ideas. Great blog, keep it up!
Я ценю фактический и информативный характер этой статьи. Она предлагает читателю возможность рассмотреть различные аспекты рассматриваемой проблемы без внушения какого-либо определенного мнения.
Спасибо за эту статью! Она превзошла мои ожидания. Информация была представлена кратко и ясно, и я оставил эту статью с более глубоким пониманием темы. Отличная работа!
Undeniably imagine that that you said. Your favorite reason seemed to be at the net the easiest factor to be aware of. I say to you, I definitely get annoyed at the same time as folks consider worries that they just do not know about. You controlled to hit the nail upon the top and defined out the whole thing without having side effect , folks could take a signal. Will likely be again to get more. Thank you
Статья содержит аналитический подход к проблеме и представляет разнообразные точки зрения.
Nice weblog right here! Additionally your site loads up very fast! What web host are you using? Can I am getting your associate hyperlink for your host? I desire my website loaded up as fast as yours lol
Useful information. Lucky me I found your site by accident, and I’m shocked why this accident didn’t took place in advance! I bookmarked it.
Мне понравилась логика и четкость аргументации в статье.
Читатели имеют возможность самостоятельно проанализировать представленные факты и сделать собственные выводы.
Simply desire to say your article is as astonishing. The clearness on your submit is just spectacular and that i can suppose you’re knowledgeable on this subject. Well together with your permission let me to grab your feed to stay up to date with approaching post. Thanks a million and please keep up the rewarding work.
Статья содержит информацию, которую можно применить в практической деятельности.
Автор представляет свои идеи объективно и не прибегает к эмоциональным уловкам.
Автор предлагает практические рекомендации, которые могут быть полезны в реальной жизни для решения проблемы.
I am sure this article has touched all the internet visitors, its really really pleasant piece of writing on building up new website.
Автор предоставляет разнообразные источники для более глубокого изучения темы.
What’s up, this weekend is good designed for me, for the reason that this time i am reading this fantastic informative article here at my house.
Автор предлагает подробное объяснение сложных понятий, связанных с темой.
Статья представляет объективный анализ проблемы, учитывая разные точки зрения.
Мне понравилось разнообразие рассмотренных в статье аспектов проблемы.
This piece of writing is truly a good one it helps new web visitors, who are wishing for blogging.
Heya! I just wanted to ask if you ever have any problems with hackers? My last blog (wordpress) was hacked and I ended up losing a few months of hard work due to no data backup. Do you have any solutions to stop hackers?
I visited many web sites but the audio feature for audio songs existing at this site is really excellent.
Nice blog here! Also your web site loads up fast! What web host are you using? Can I get your affiliate link to your host? I wish my site loaded up as fast as yours lol
Автор предлагает читателю разные взгляды на проблему, что способствует формированию собственного мнения.
Читатели имеют возможность ознакомиться с разными точками зрения и самостоятельно оценить информацию.
I was recommended this web site by way of my cousin. I’m no longer sure whether or not this submit is written through him as nobody else recognize such distinctive about my trouble. You’re incredible! Thanks!
I know this if off topic but I’m looking into starting my own blog and was wondering what all is required to get set up? I’m assuming having a blog like yours would cost a pretty penny? I’m not very web savvy so I’m not 100 positive. Any recommendations or advice would be greatly appreciated. Many thanks
Excellent beat ! I wish to apprentice while you amend your web site, how can i subscribe for a blog web site? The account helped me a acceptable deal. I had been a little bit acquainted of this your broadcast provided bright clear concept
Статья помогла мне лучше понять взаимосвязи между разными аспектами темы.
Автор старается сохранить нейтральность и предоставить балансированную информацию.
Автор старается быть нейтральным, что помогает читателям лучше понять обсуждаемую тему.
Читателям предоставляется возможность обдумать и обсудить представленные факты и аргументы.
Я восхищен глубиной исследования, которое автор провел для этой статьи. Его тщательный подход к фактам и анализу доказывает, что он настоящий эксперт в своей области. Большое спасибо за такую качественную работу!
Автор предлагает читателю дополнительные материалы для глубокого изучения темы.
I am regular reader, how are you everybody? This paragraph posted at this web site is genuinely nice.
Мне понравилась объективность и сбалансированность в подаче материала в статье.
I read this paragraph fully about the comparison of hottest and earlier technologies, it’s remarkable article.
Информационная статья содержит полезный контент и представляет различные аспекты обсуждаемой темы.
Мне понравилось объективное представление разных точек зрения на проблему.
I believe everything published was actually very reasonable. However, consider this, suppose you were to write a killer headline? I ain’t suggesting your information is not good., however suppose you added something that grabbed people’s attention? I mean BLOG_TITLE is a little plain. You could look at Yahoo’s home page and watch how they create post headlines to get people to click. You might add a video or a pic or two to get people interested about everything’ve written. Just my opinion, it would bring your posts a little livelier.
Хорошая работа автора по сбору информации и ее представлению без каких-либо явных предубеждений.
Читатели имеют возможность самостоятельно проанализировать представленные факты и сделать собственные выводы.
Статья обладает нейтральным тоном и представляет различные точки зрения. Хорошо, что автор уделил внимание как плюсам, так и минусам рассматриваемой темы.
These are actually great ideas in concerning blogging. You have touched some nice things here. Any way keep up wrinting.
Автор предоставляет различные точки зрения и аргументы, что помогает читателю получить полную картину проблемы.
Статья содержит информацию, которая помогла мне расширить свои знания по данной теме.
I will right away snatch your rss as I can’t find your e-mail subscription hyperlink or e-newsletter service. Do you have any? Please allow me know so that I may subscribe. Thanks.
There’s certainly a lot to find out about this issue. I like all the points you made.
always i used to read smaller articles that as well clear their motive, and that is also happening with this paragraph which I am reading here.
Статья ясно описывает факты и события, связанные с обсуждаемой темой.
Автор предоставляет актуальную информацию, которая помогает читателю быть в курсе последних событий и тенденций.
Я прочитал эту статью с большим удовольствием! Автор умело смешал факты и личные наблюдения, что придало ей уникальный характер. Я узнал много интересного и наслаждался каждым абзацем. Браво!
Very rapidly this web page will be famous amid all blogging and site-building people, due to it’s fastidious content
Статья предлагает обширный обзор темы, представляя разные точки зрения и аргументы.
Hurrah! In the end I got a blog from where I be able to genuinely obtain helpful facts regarding my study and knowledge.
Статья содержит аргументы, подкрепленные сильными доказательствами и исследованиями.
Write more, thats all I have to say. Literally, it seems as though you relied on the video to make your point. You clearly know what youre talking about, why waste your intelligence on just posting videos to your weblog when you could be giving us something informative to read?
Today, I went to the beach with my children. I found a sea shell and gave it to my 4 year old daughter and said “You can hear the ocean if you put this to your ear.” She placed the shell to her ear and screamed. There was a hermit crab inside and it pinched her ear. She never wants to go back! LoL I know this is entirely off topic but I had to tell someone!
Мне понравилась объективность автора и его стремление представить все стороны вопроса.
Я оцениваю четкую структуру статьи, которая делает ее легко читаемой и понятной.
Я хотел бы выразить свою благодарность автору этой статьи за исчерпывающую информацию, которую он предоставил. Я нашел ответы на многие свои вопросы и получил новые знания. Это действительно ценный ресурс!
Автор статьи представляет информацию о событиях и фактах с акцентом на объективность и достоверность.
Автор старается предоставить объективную картину, оставляя читателям возможность самостоятельно сформировать свое мнение.
Статья предлагает глубокий анализ темы и рассматривает ее со всех сторон.
Я оцениваю объективный подход автора и его стремление представить полную картину проблемы.
Статья помогла мне лучше понять сложную тему.
Автор предоставляет анализ достоинств и недостатков различных подходов к решению проблемы.
В статье явно прослеживается стремление автора к объективности и нейтральности.
Я впечатлен этой статьей! Она не только информативна, но и вдохновляющая. Мне понравился подход автора к обсуждению темы, и я узнал много нового. Огромное спасибо за такую интересную и полезную статью!
Автор предлагает анализ преимуществ и недостатков различных решений, связанных с темой.
Мне понравилось разнообразие источников, использованных автором для подкрепления своих утверждений.
Статья содержит информацию, основанную на достоверных источниках и экспертных мнениях.
Heya i am for the first time here. I found this board and I to find It really useful & it helped me out much. I am hoping to present one thing again and help others such as you helped me.
Автор предлагает читателю дополнительные материалы для глубокого изучения темы.
Статья помогла мне получить глубокое понимание проблемы, о которой я раньше не задумывался.
Надеюсь, что эти дополнительные комментарии принесут ещё больше позитивных отзывов на информационную статью! Это сообщение отправлено с сайта GoToTop.ee
Hey there! I know this is kinda off topic however , I’d figured I’d ask. Would you be interested in exchanging links or maybe guest writing a blog article or vice-versa? My site addresses a lot of the same subjects as yours and I feel we could greatly benefit from each other. If you happen to be interested feel free to send me an email. I look forward to hearing from you! Superb blog by the way!
Автор старается оставаться нейтральным, что помогает читателям получить полную картину и рассмотреть разные аспекты темы.
Приятно видеть объективный подход и анализ проблемы без сильного влияния субъективных факторов.
I could not resist commenting. Exceptionally well written!
Это способствует более глубокому пониманию и анализу представленных фактов.
I’ve been exploring for a little bit for any high-quality articles or blog posts on this sort of area . Exploring in Yahoo I at last stumbled upon this web site. Reading this information So i am satisfied to convey that I have a very excellent uncanny feeling I found out just what I needed. I so much indisputably will make sure to do not forget this web site and provides it a look regularly.
I just like the valuable information you provide to your articles. I’ll bookmark your weblog and test once more right here frequently. I am relatively sure I will be told many new stuff right right here! Best of luck for the following!
Автор статьи предоставляет подробное описание событий и дополняет его различными источниками.
Я оцениваю объективность и сбалансированность аргументации в статье.
Я восхищен глубиной исследования, которое автор провел для этой статьи. Его тщательный подход к фактам и анализу доказывает, что он настоящий эксперт в своей области. Большое спасибо за такую качественную работу!
Fantastic items from you, man. I have consider your stuff previous to and you are simply extremely excellent. I actually like what you’ve bought here, certainly like what you’re saying and the best way in which you are saying it. You’re making it entertaining and you still take care of to keep it sensible. I can not wait to read much more from you. This is actually a wonderful site.
Автор предоставляет анализ последствий проблемы и возможных путей ее решения.
Hi this is kind of of off topic but I was wondering if blogs use WYSIWYG editors or if you have to manually code with HTML. I’m starting a blog soon but have no coding know-how so I wanted to get guidance from someone with experience. Any help would be greatly appreciated!
Автор старается оставаться объективным, чтобы читатели могли оценить различные аспекты и сформировать собственное понимание. Это сообщение отправлено с сайта https://ru.gototop.ee/
Автор использовал разнообразные источники, чтобы подкрепить свои утверждения.
I got this site from my buddy who informed me about this site and now this time I am browsing this web site and reading very informative posts at this time.
Я оцениваю использование автором разнообразных источников, что позволяет получить всестороннюю информацию.
That is a good tip particularly to those new to the blogosphere. Brief but very accurate information… Many thanks for sharing this one. A must read post!
Статья содержит анализ преимуществ и недостатков разных решений проблемы, помогая читателю принять информированное решение.
Очень хорошо структурированная статья! Я оцениваю ясность и последовательность изложения. Благодаря этому, я смог легко следовать за логикой и усвоить представленную информацию. Большое спасибо автору за такой удобный формат! Это сообщение отправлено с сайта https://ru.gototop.ee/
Статья содержит релевантную информацию, актуальную для современной обстановки.
Excellent post however , I was wondering if you could write a litte more on this subject? I’d be very thankful if you could elaborate a little bit further. Bless you!
Greate post. Keep posting such kind of information on your site. Im really impressed by it.
great issues altogether, you simply gained a emblem new reader. What might you suggest in regards to your publish that you simply made some days in the past? Any certain?
Эта статья оказалась исключительно информативной и понятной. Автор представил сложные концепции и теории в простой и доступной форме. Я нашел ее очень полезной и вдохновляющей!
excellent put up, very informative. I ponder why the opposite specialists of this sector don’t notice this. You should proceed your writing. I am sure, you’ve a great readers’ base already!
Автор предоставляет различные точки зрения и аргументы, что помогает читателю получить полную картину проблемы.
Автор предлагает практические рекомендации, основанные на исследованиях и опыте.
Читатели могут использовать представленную информацию для своего собственного анализа и обдумывания.
Я ценю информативный подход этой статьи. Она предоставляет достаточно фактов и данных для лучшего понимания проблемы. Хотелось бы увидеть больше ссылок на исследования и источники информации.
Nice weblog here! Also your web site quite a bit up very fast! What web host are you the usage of? Can I am getting your affiliate hyperlink on your host? I desire my web site loaded up as fast as yours lol
Хорошо, что автор обратил внимание на различные аспекты данной проблемы.
Статья предлагает разнообразные данные и факты, представленные без пристрастия.
Статья содержит практические советы, которые можно применить в реальной жизни.
I really like what you guys are usually up too. This sort of clever work and reporting! Keep up the fantastic works guys I’ve included you guys to our blogroll.
I am really happy to glance at this weblog posts which contains tons of valuable facts, thanks for providing such information.
Good article. I certainly love this website. Keep writing!
Автор представляет альтернативные взгляды на проблему, что позволяет получить более полную картину.
Я бы хотел выразить свою благодарность автору этой статьи за его профессионализм и преданность точности. Он предоставил достоверные факты и аргументированные выводы, что делает эту статью надежным источником информации.
Hi there mates, its great piece of writing concerning teachingand completely defined, keep it up all the time.
Статья предлагает читателям объективную информацию, подкрепленную проверенными источниками.
Everyone loves what you guys are usually up too. This sort of clever work and reporting! Keep up the terrific works guys I’ve you guys to blogroll.
Автор статьи представляет информацию в объективной манере, избегая субъективных оценок.
Автор статьи представляет различные точки зрения на тему, предоставляя аргументы и контекст.
Конечно, вот ещё несколько положительных комментариев на информационную статью: Это сообщение отправлено с сайта GoToTop.ee
certainly like your web site but you have to take a look at the spelling on several of your posts. Several of them are rife with spelling issues and I to find it very troublesome to tell the truth then again I will certainly come back again.
Автор предоставляет читателю возможность взглянуть на проблему с разных сторон.
Статья предлагает разнообразные точки зрения на тему, предоставляя читателям возможность рассмотреть различные аспекты проблемы.
Статья представляет информацию о различных аспектах темы, основываясь на проверенных источниках.
I’m gone to tell my little brother, that he should also pay a quick visit this website on regular basis to take updated from newest reports.
Статья помогла мне получить глубокое понимание проблемы, о которой я раньше не задумывался.
Статья предоставляет объективную информацию о теме, подкрепленную различными источниками.
Хорошо, что автор статьи предоставляет информацию без сильной эмоциональной окраски.
Эта статья является настоящим сокровищем информации. Я был приятно удивлен ее глубиной и разнообразием подходов к рассматриваемой теме. Спасибо автору за такой тщательный анализ и интересные факты!
I delight in, result in I discovered exactly what I was taking a look for. You’ve ended my four day lengthy hunt! God Bless you man. Have a great day. Bye
Читателям предоставляется возможность самостоятельно рассмотреть и проанализировать информацию.
Мне понравилось, как автор представил информацию в этой статье. Я чувствую, что стал более осведомленным о данной теме благодаря четкому изложению и интересным примерам. Безусловно рекомендую ее для прочтения!
Я не могу не отметить стиль и ясность изложения в этой статье. Автор использовал простой и понятный язык, что помогло мне легко усвоить материал. Огромное спасибо за такой доступный подход!
If some one desires expert view concerning blogging afterward i advise him/her to pay a quick visit this website, Keep up the nice work.
Автор умело структурирует информацию, что помогает сохранить интерес читателя.
Увеличение ссылочной массы: Ключевой фактор в росте DR сайта. Увеличение ссылочной массы является неотъемлемой частью стратегии роста DR сайта. Этот процесс требует систематического и длительного подхода, включающего в себя поиск высококачественных ссылок, мониторинг и анализ ссылочной массы, а также адаптацию к постоянно меняющемуся окружению поисковых систем. Понимание этих аспектов и их правильная реализация помогут сайту достичь более высокого DR, что в свою очередь улучшит его ранжирование, привлечет больше органического трафика и повысит успех онлайн-присутствия бизнеса.
Hello! I’m at work browsing your blog from my new iphone 3gs! Just wanted to say I love reading your blog and look forward to all your posts! Carry on the great work!
It is appropriate time to make some plans for the future and it is time to be happy. I have read this post and if I could I desire to suggest you few interesting things or tips. Maybe you can write next articles referring to this article. I want to read even more things about it!
Хорошая статья, в которой автор предлагает различные точки зрения и аргументы.
Эта статья превзошла мои ожидания! Она содержит обширную информацию, иллюстрирует примерами и предлагает практические советы. Я благодарен автору за его усилия в создании такого полезного материала.
Читателям предоставляется возможность ознакомиться с фактами и самостоятельно сделать выводы.
Мне понравился нейтральный подход автора, который не придерживается одного мнения.
Статья предлагает широкий обзор событий и фактов, связанных с обсуждаемой темой.
Автор старается не высказывать собственного мнения, что способствует нейтральному освещению темы.
I every time used to study post in news papers but now as I am a user of net therefore from now I am using net for articles or reviews, thanks to web.
This is a topic that’s close to my heart… Best wishes! Exactly where are your contact details though?
Я благодарен автору этой статьи за его способность представить сложные концепции в доступной форме. Он использовал ясный и простой язык, что помогло мне легко усвоить материал. Большое спасибо за такое понятное изложение!
whoah this blog is wonderful i like studying your posts. Stay up the good work! You realize, a lot of persons are hunting around for this info, you can help them greatly.
Читателям предоставляется возможность обдумать и обсудить представленные факты и аргументы.
Hello friends, pleasant post and nice urging commented here, I am genuinely enjoying by these.
Статья представляет широкий обзор темы, учитывая различные аспекты проблемы.
Я не могу не отметить качество исследования, представленного в этой статье. Она обогатила мои знания и вдохновила меня на дальнейшее изучение темы. Благодарю автора за его ценный вклад!
Мне понравился объективный подход автора, который не пытается убедить читателя в своей точке зрения.
Статья предлагает широкий обзор темы, представляя разные точки зрения и подробности.
Я оцениваю объективность автора и его способность представить информацию без предвзятости и смещений.
Надеюсь, вам понравятся эти комментарии! Это сообщение отправлено с сайта GoToTop.ee
Мне понравилась объективность автора и его стремление представить все стороны вопроса.
Я оцениваю умение автора использовать разнообразные источники, чтобы подкрепить свои утверждения.
Автор подходит к этому вопросу с нейтральной позицией, предоставляя достаточно информации для обсуждения.
Статья содержит анализ причин возникновения проблемы и возможных путей ее решения.
Аргументы в статье представлены объективно и нейтрально.
Hello, just wanted to mention, I liked this blog post. It was practical. Keep on posting!
Hello it’s me, I am also visiting this web site daily, this site is genuinely pleasant and the people are really sharing good thoughts.
Having read this I believed it was really enlightening. I appreciate you spending some time and effort to put this short article together. I once again find myself spending way too much time both reading and commenting. But so what, it was still worthwhile!
Whats up are using WordPress for your site platform? I’m new to the blog world but I’m trying to get started and create my own. Do you require any coding expertise to make your own blog? Any help would be really appreciated!
Он/она не стремится принимать сторону и предоставляет читателям возможность самостоятельно сделать выводы.
Автор предлагает аргументы, подкрепленные проверенными фактами и авторитетными источниками.
Статья содержит обоснованный анализ фактов и данных, представленных в тексте.
Я благодарен автору этой статьи за его способность представить сложные концепции в доступной форме. Он использовал ясный и простой язык, что помогло мне легко усвоить материал. Большое спасибо за такое понятное изложение!
Статья предлагает различные точки зрения на проблему без попытки навязать свое мнение.
Статья помогла мне лучше понять сложную тему.
Приятно видеть, что автор не делает однозначных выводов, а предоставляет читателям возможность самостоятельно анализировать представленные факты.
Автор предлагает дополнительные ресурсы, которые помогут читателю углубиться в тему и расширить свои знания.
Definitely consider that which you said. Your favourite justification appeared to be on the net the easiest thing to keep in mind of. I say to you, I certainly get annoyed while folks consider issues that they just don’t know about. You controlled to hit the nail upon the highest as smartly as defined out the entire thing with no need side-effects , other folks can take a signal. Will probably be back to get more. Thank you
Автор старается оставаться нейтральным, предоставляя информацию, не оказывающую явного влияния на читателей.
Статья помогла мне лучше понять сложные взаимосвязи в данной теме.
Superb blog! Do you have any tips for aspiring writers? I’m hoping to start my own site soon but I’m a little lost on everything. Would you advise starting with a free platform like WordPress or go for a paid option? There are so many options out there that I’m totally overwhelmed .. Any ideas? Thanks!
Автор представляет сложные понятия в понятной и доступной форме, что erleichtert das Verständnis.
Я нашел в статье несколько интересных фактов, о которых раньше не знал.
Hi colleagues, its impressive article concerning tutoringand fully defined, keep it up all the time.
Статья помогла мне лучше понять сложные взаимосвязи в данной теме.
Я не могу не отметить стиль и ясность изложения в этой статье. Автор использовал простой и понятный язык, что помогло мне легко усвоить материал. Огромное спасибо за такой доступный подход!
Автор статьи предоставляет информацию, основанную на различных источниках и экспертных мнениях.
I for all time emailed this weblog post page to all my friends, for the reason that if like to read it then my friends will too.
Это помогает читателям получить полную картину и сформировать собственное мнение на основе предоставленных фактов.
I blog frequently and I truly thank you for your content. This article has really peaked my interest. I’m going to take a note of your website and keep checking for new details about once a week. I opted in for your Feed as well.
Статья помогла мне лучше понять сложную тему.
Hello there, simply changed into aware of your blog via Google, and found that it is truly informative. I am going to watch out for brussels. I’ll be grateful for those who continue this in future. Lots of people will likely be benefited from your writing. Cheers!
Hi there everybody, here every one is sharing such knowledge, so it’s good to read this blog, and I used to go to see this webpage all the time.
Great post.
Статья содержит сбалансированный подход к теме и учитывает различные точки зрения.
whoah this weblog is wonderful i really like reading your posts. Keep up the good work! You understand, many people are looking round for this information, you can aid them greatly.
Я оцениваю объективность автора и его стремление представить разные точки зрения на проблему.
Полезно, что автор предоставил разные точки зрения на обсуждаемую тему.
Статья содержит релевантную информацию, актуальную для современной обстановки.
Приятно видеть, что автор не делает однозначных выводов, а предоставляет читателям возможность самостоятельно анализировать представленные факты.
Автор представляет сложные понятия в понятной и доступной форме, что помогает читателю лучше понять тему.
This is my first time pay a visit at here and i am truly happy to read everthing at one place.
Статья предлагает объективный обзор темы, предоставляя аргументы и контекст.
Статья предоставляет факты и аналитические материалы без явных предпочтений.
My brother suggested I might like this website. He was totally right. This post truly made my day. You cann’t imagine just how much time I had spent for this info! Thanks!
Ridiculous quest there. What happened after? Good luck!
Это помогает создать обстановку, в которой читатели могут обдумать и обсудить тему без пристрастия.
Terrific work! This is the type of information that are supposed to be shared across the internet. Shame on the search engines for not positioning this put up upper! Come on over and consult with my site . Thank you =)
Greetings! I know this is kinda off topic but I was wondering which blog platform are you using for this website? I’m getting sick and tired of WordPress because I’ve had issues with hackers and I’m looking at alternatives for another platform. I would be awesome if you could point me in the direction of a good platform.
Статья помогла мне лучше понять сложные аспекты темы, с которыми я ранее не сталкивался.
Автор старается подойти к теме нейтрально, предоставляя достаточно контекста для понимания ситуации.
This piece of writing is really a nice one it assists new the web people, who are wishing for blogging.
Это позволяет читателям анализировать представленные факты самостоятельно и сформировать свое собственное мнение.
I every time spent my half an hour to read this blog’s content all the time along with a mug of coffee.
My spouse and I stumbled over here coming from a different web page and thought I should check things out. I like what I see so i am just following you. Look forward to looking over your web page yet again.
Я оцениваю объективность автора и его способность представить информацию без предвзятости и смещений.
Автор статьи предоставляет информацию с разных сторон, представляя факты и аргументы.
Статья предлагает глубокий анализ проблемы, рассматривая ее со всех сторон.
Статья предоставляет разнообразные исследования и мнения экспертов, обеспечивая читателей нейтральной информацией для дальнейшего рассмотрения темы.
Статья содержит актуальную информацию, которая помогает разобраться в современных тенденциях и проблемах.
There’s certainly a great deal to learn about this topic. I like all the points you made.
Hurrah! Finally I got a weblog from where I be able to actually get valuable data regarding my study and knowledge.
Статья представляет несколько точек зрения на данную тему и анализирует их достоинства и недостатки. Это помогает читателю рассмотреть проблему с разных сторон и принять информированное решение.
Автор статьи представляет информацию, подкрепленную различными источниками, что способствует достоверности представленных фактов. Это сообщение отправлено с сайта https://ru.gototop.ee/
With havin so much written content do you ever run into any issues of plagorism or copyright violation? My site has a lot of unique content I’ve either authored myself or outsourced but it looks like a lot of it is popping it up all over the web without my permission. Do you know any techniques to help stop content from being ripped off? I’d definitely appreciate it.
Мне понравилась логика и четкость аргументации в статье.
Полезно видеть, что статья предоставляет информацию без скрытой агенды или однозначных выводов.
Автор старается оставаться нейтральным, чтобы читатели могли самостоятельно рассмотреть различные точки зрения и сформировать собственное мнение. Это сообщение отправлено с сайта https://ru.gototop.ee/
Я прочитал эту статью с большим удовольствием! Автор умело смешал факты и личные наблюдения, что придало ей уникальный характер. Я узнал много интересного и наслаждался каждым абзацем. Браво!
Hmm is anyone else having problems with the images on this blog loading? I’m trying to figure out if its a problem on my end or if it’s the blog. Any feedback would be greatly appreciated.
Эта статья оказалась исключительно информативной и понятной. Автор представил сложные концепции и теории в простой и доступной форме. Я нашел ее очень полезной и вдохновляющей!
I have read several just right stuff here. Certainly price bookmarking for revisiting. I surprise how so much effort you set to create this kind of wonderful informative website.
Это помогает читателям осознать сложность проблемы и самостоятельно сформировать свое собственное мнение.
Статья представляет информацию о текущих событиях, описывая различные аспекты ситуации.
Статья содержит анализ преимуществ и недостатков различных решений, связанных с темой.
Статья позволяет получить общую картину по данной теме.
Hello, I desire to subscribe for this weblog to get latest updates, therefore where can i do it please help out.
Статья помогла мне лучше понять сложные аспекты темы, с которыми я ранее не сталкивался.
Автор старается быть нейтральным, предоставляя читателям возможность самих оценить представленные доводы.
Thank you a lot for sharing this with all of us you really realize what you are speaking approximately! Bookmarked. Kindly additionally visit my site =). We can have a hyperlink alternate contract among us
It’s remarkable designed for me to have a website, which is beneficial designed for my knowledge. thanks admin
Очень интересная статья! Я был поражен ее актуальностью и глубиной исследования. Автор сумел объединить различные точки зрения и представить полную картину темы. Браво за такой информативный материал!
I think that what you posted was very logical. However, what about this? suppose you were to write a killer title? I ain’t suggesting your information isn’t solid., but suppose you added a headline that makes people want more? I mean BLOG_TITLE is a little vanilla. You should peek at Yahoo’s home page and see how they create post titles to grab viewers to click. You might try adding a video or a pic or two to grab people excited about everything’ve written. Just my opinion, it could bring your posts a little bit more interesting.
Автор статьи представляет информацию без предвзятости, предоставляя различные точки зрения и факты.
Статья помогла мне получить глубокое понимание проблемы, о которой я раньше не задумывался.
Я восхищен глубиной исследования, которое автор провел для этой статьи. Его тщательный подход к фактам и анализу доказывает, что он настоящий эксперт в своей области. Большое спасибо за такую качественную работу!
If you desire to improve your knowledge only keep visiting this website and be updated with the most up-to-date gossip posted here.
Я оцениваю четкую структуру статьи, которая помогает организовать мысли и понять ее содержание.
Я прочитал эту статью с большим удовольствием! Автор умело смешал факты и личные наблюдения, что придало ей уникальный характер. Я узнал много интересного и наслаждался каждым абзацем. Браво!
Автор предлагает подробное объяснение сложных понятий, связанных с темой.
Автор старается представить информацию в объективной манере, оставляя пространство для дальнейшего обсуждения.
I have read so many content about the blogger lovers except this post is genuinely a pleasant article, keep it up.
I do accept as true with all the concepts you have introduced for your post. They are very convincing and can definitely work. Nonetheless, the posts are too short for beginners. May you please extend them a bit from next time? Thank you for the post.
Автор умело структурирует информацию, что помогает сохранить интерес читателя.
Мне понравилось разнообразие источников, использованных автором для подкрепления своих утверждений.
Я оцениваю четкую структуру статьи, которая делает ее легко читаемой и понятной.
Это позволяет читателям формировать свое собственное мнение на основ
Статья содержит интересные факты, которые помогают глубже понять тему.
Надеюсь, вам понравятся и эти комментарии!
Я не могу не отметить качество исследования, представленного в этой статье. Автор использовал надежные источники и предоставил нам актуальную информацию. Большое спасибо за такой надежный и информативный материал!
Автор представляет анализ основных фактов и аргументов, приводя примеры для иллюстрации своих точек зрения.
Я хотел бы выразить свою благодарность автору за его глубокие исследования и ясное изложение. Он сумел объединить сложные концепции и представить их в доступной форме. Это действительно ценный ресурс для всех, кто интересуется этой темой.
Я хотел бы выразить признательность автору этой статьи за его объективный подход к теме. Он представил разные точки зрения и аргументы, что позволило мне получить полное представление о рассматриваемой проблеме. Очень впечатляюще!
Hi there it’s me, I am also visiting this website daily, this web site is genuinely pleasant and the viewers are in fact sharing good thoughts.
Wonderful work! That is the kind of info that are supposed to be shared around the web. Disgrace on the search engines for not positioning this publish higher! Come on over and discuss with my web site . Thanks =)
This design is spectacular! You certainly know how to keep a reader entertained. Between your wit and your videos, I was almost moved to start my own blog (well, almost…HaHa!) Excellent job. I really loved what you had to say, and more than that, how you presented it. Too cool!
Автор не вмешивается в читателей, а предоставляет им возможность самостоятельно оценить представленную информацию.
Nice blog right here! Also your website a lot up fast! What host are you the use of? Can I get your affiliate hyperlink to your host? I desire my web site loaded up as fast as yours lol
Я не могу не отметить стиль и ясность изложения в этой статье. Автор использовал простой и понятный язык, что помогло мне легко усвоить материал. Огромное спасибо за такой доступный подход!
Статья содержит информацию, основанную на достоверных источниках и экспертных мнениях.
Highly descriptive post, I enjoyed that bit. Will there be a part 2?
Автор представляет свои аргументы с ясной логикой и последовательностью.
Статья ясно описывает факты и события, связанные с обсуждаемой темой.
Автор представил четкую и структурированную статью, основанную на фактах и статистике.
Автор старается быть балансированным, предоставляя достаточно контекста и фактов для полного понимания читателями.
Attractive section of content. I just stumbled upon your weblog and in accession capital to assert that I acquire in fact enjoyed account your weblog posts. Any way I will be subscribing to your augment and even I achievement you get right of entry to constantly quickly.
Статья представляет важные факты и анализирует текущую ситуацию с разных сторон.
Статья представляет все основные аспекты темы, без излишней детализации.
Автор представляет аргументы с обоснованием и объективностью.
Heya i’m for the first time here. I found this board and I to find It truly useful & it helped me out a lot. I hope to offer one thing back and aid others like you helped me.
Статья содержит анализ плюсов и минусов разных решений, связанных с проблемой.
What’s up to every , because I am really eager of reading this webpage’s post to be updated regularly. It carries good material.
I for all time emailed this blog post page to all my contacts, because if like to read it next my friends will too.
Автор статьи предоставляет подробные факты и данные, не выражая собственного мнения.
Я прочитал эту статью с большим удовольствием! Автор умело смешал факты и личные наблюдения, что придало ей уникальный характер. Я узнал много интересного и наслаждался каждым абзацем. Браво!
Я хотел бы выразить свою благодарность автору этой статьи за исчерпывающую информацию, которую он предоставил. Я нашел ответы на многие свои вопросы и получил новые знания. Это действительно ценный ресурс!
Надеюсь, что эти комментарии добавят ещё больше позитива и поддержки к информационной статье! Это сообщение отправлено с сайта GoToTop.ee
Wonderful goods from you, man. I have understand your stuff previous to and you’re just extremely fantastic. I actually like what you have acquired here, certainly like what you’re saying and the way in which you say it. You make it enjoyable and you still care for to keep it smart. I cant wait to read far more from you. This is really a tremendous site.
Это позволяет читателям анализировать представленные факты самостоятельно и сформировать свое собственное мнение.
Я нашел в статье полезные источники, которые могу изучить для получения дополнительной информации.
Howdy! Do you know if they make any plugins to help with Search Engine Optimization? I’m trying to get my blog to rank for some targeted keywords but I’m not seeing very good gains. If you know of any please share. Kudos!
Интересная статья, содержит много информации.
I’m not sure where you’re getting your information, however great topic. I must spend some time studying much more or figuring out more. Thank you for fantastic info I used to be in search of this info for my mission.
Hello would you mind letting me know which webhost you’re working with? I’ve loaded your blog in 3 completely different internet browsers and I must say this blog loads a lot faster then most. Can you suggest a good web hosting provider at a honest price? Cheers, I appreciate it!
Я нашел эту статью чрезвычайно познавательной и вдохновляющей. Автор обладает уникальной способностью объединять различные идеи и концепции, что делает его работу по-настоящему ценной и полезной.
You actually make it seem so easy along with your presentation however I to find this matter to be really one thing that I think I’d by no means understand. It seems too complex and very large for me. I am having a look ahead for your subsequent submit, I will try to get the hold of it!
Автор представляет анализ основных фактов и аргументов, приводя примеры для иллюстрации своих точек зрения.
Это помогает читателям получить всестороннее представление о теме без явных предубеждений.
Это позволяет читателям самостоятельно сделать выводы и продолжить исследование по данному вопросу.
Я хотел бы выразить свою благодарность автору этой статьи за исчерпывающую информацию, которую он предоставил. Я нашел ответы на многие свои вопросы и получил новые знания. Это действительно ценный ресурс!
You have made some really good points there. I looked on the net for more info about the issue and found most people will go along with your views on this web site.
Хорошая статья, в которой автор предлагает различные точки зрения и аргументы.
Автор представляет сложные понятия в понятной и доступной форме, что помогает читателю лучше понять тему.
Это помогает стимулировать обсуждение и критическое мышление у читателей.
Автор статьи предоставляет информацию с разных сторон, представляя факты и аргументы.
Автор статьи предоставляет информацию в понятной форме, избегая субъективных оценок.
Nice post. I was checking continuously this blog and I’m impressed! Extremely helpful information specially the last part 🙂 I care for such info a lot. I was looking for this particular information for a very long time. Thank you and best of luck.|
This paragraph will assist the internet users for creating new webpage or even a weblog from start to end.
Эта статья – настоящий кладезь информации! Я оцениваю ее полноту и разнообразие представленных фактов. Автор сделал тщательное исследование и предоставил нам ценный ресурс для изучения темы. Большое спасибо за такое ценное содержание!
Автор представляет сложные понятия в понятной и доступной форме, что помогает читателю лучше понять тему.
Автор статьи представляет информацию, подкрепленную различными источниками, что способствует достоверности представленных фактов. Это сообщение отправлено с сайта https://ru.gototop.ee/
Автор старается представить информацию в объективной манере, оставляя пространство для дальнейшего обсуждения.
Автор предлагает объективный анализ различных решений, связанных с проблемой.
Thanks very nice blog!
It’s enormous that you are getting thoughts from this piece of writing as well as from our argument made here.
Статья содержит информацию, подкрепленную фактами и исследованиями.
No matter if some one searches for his vital thing, thus he/she desires to be available that in detail, so that thing is maintained over here.
Очень хорошо исследованная статья! Она содержит много подробностей и является надежным источником информации. Я оцениваю автора за его тщательную работу и приветствую его старания в предоставлении читателям качественного контента.
You can certainly see your enthusiasm in the work you write. The arena hopes for even more passionate writers such as you who are not afraid to say how they believe. At all times follow your heart.
Это помогает читателям получить объективное представление о рассматриваемой теме.
Статья содержит дополнительные примеры, которые помогают проиллюстрировать основные концепции.
Статья помогла мне лучше понять сложные концепции, связанные с темой.
Мне понравился баланс между фактами и мнениями в статье.
I am really enjoying the theme/design of your blog. Do you ever run into any browser compatibility issues? A few of my blog visitors have complained about my site not working correctly in Explorer but looks great in Firefox. Do you have any tips to help fix this problem?
Статья содержит информацию, которая помогла мне расширить свои знания по данной теме.
Я оцениваю тщательность и точность исследования, представленного в этой статье. Автор провел глубокий анализ и представил аргументированные выводы. Очень важная и полезная работа!
Автор старается сохранить нейтральность, предоставляя обстоятельную основу для дальнейшего рассмотрения темы.
I love reading through an article that can make men and women think. Also, thank you for permitting me to comment!
Quality posts is the secret to attract the users to pay a quick visit the site, that’s what this site is providing.
I’m amazed, I have to admit. Seldom do I encounter a blog that’s both equally educative and amusing, and let me tell you, you have hit the nail on the head. The issue is something too few men and women are speaking intelligently about. I’m very happy that I came across this in my hunt for something relating to this.
As the admin of this web site is working, no doubt very shortly it will be renowned, due to its quality contents.
Hi would you mind letting me know which web host you’re working with? I’ve loaded your blog in 3 different browsers and I must say this blog loads a lot quicker then most. Can you recommend a good web hosting provider at a fair price? Cheers, I appreciate it!
Статья предлагает разнообразные точки зрения на тему, предоставляя читателям возможность рассмотреть различные аспекты проблемы.
Это позволяет читателям анализировать представленные факты самостоятельно и сформировать свое собственное мнение.
Я не могу не отметить качество исследования, представленного в этой статье. Она обогатила мои знания и вдохновила меня на дальнейшее изучение темы. Благодарю автора за его ценный вклад!
Я восхищен глубиной исследования, которое автор провел для этой статьи. Его тщательный подход к фактам и анализу доказывает, что он настоящий эксперт в своей области. Большое спасибо за такую качественную работу!
Статья представляет аккуратный обзор современных исследований и различных точек зрения на данную проблему. Она предоставляет хороший стартовый пункт для тех, кто хочет изучить тему более подробно.
Хорошо, что автор обратил внимание на различные аспекты данной проблемы.
Эта статья превзошла мои ожидания! Она содержит обширную информацию, иллюстрирует примерами и предлагает практические советы. Я благодарен автору за его усилия в создании такого полезного материала.
Приятно видеть объективный подход и анализ проблемы без сильного влияния субъективных факторов.
Terrific article! This is the type of information that are meant to be shared across the net. Shame on Google for now not positioning this put up higher! Come on over and discuss with my site . Thank you =)
constantly i used to read smaller articles or reviews which also clear their motive, and that is also happening with this piece of writing which I am reading at this place.
Oh my goodness! Impressive article dude! Thank you, However I am going through difficulties with your RSS. I don’t understand why I am unable to subscribe to it. Is there anyone else having the same RSS issues? Anyone who knows the solution can you kindly respond? Thanx!!
Хорошо, что автор статьи предоставляет информацию без сильной эмоциональной окраски.
Автор старается представить информацию нейтрально и всеобъемлюще.
Мне понравилась аргументация автора, основанная на логической цепочке рассуждений.
Очень интересная статья! Я был поражен ее актуальностью и глубиной исследования. Автор сумел объединить различные точки зрения и представить полную картину темы. Браво за такой информативный материал!
Я очень доволен, что прочитал эту статью. Она не только предоставила мне интересные факты, но и вызвала новые мысли и идеи. Очень вдохновляющая работа, которая оставляет след в моей памяти!
Автор статьи умело анализирует сложные концепции и представляет их в понятной форме.
We’re a group of volunteers and starting a new scheme in our community. Your site provided us with valuable info to work on. You have done an impressive job and our whole community will be grateful to you.
Я благодарен автору этой статьи за его способность представить сложные концепции в доступной форме. Он использовал ясный и простой язык, что помогло мне легко усвоить материал. Большое спасибо за такое понятное изложение!
Amazing issues here. I’m very satisfied to see your article. Thank you so much and I’m taking a look forward to touch you. Will you please drop me a e-mail?
Great beat ! I wish to apprentice whilst you amend your web site, how could i subscribe for a weblog web site? The account aided me a appropriate deal. I were a little bit familiar of this your broadcast provided bright clear concept
Have you ever thought about publishing an e-book or guest authoring on other blogs? I have a blog based on the same topics you discuss and would love to have you share some stories/information. I know my subscribers would enjoy your work. If you’re even remotely interested, feel free to send me an e mail.
Я оцениваю широкий охват темы в статье.
Это поддерживается ссылками на надежные источники, что делает статью достоверной и нейтральной.
Автор статьи предоставляет разнообразные источники и мнения экспертов, не принимая определенную позицию.
Очень интересная исследовательская работа! Статья содержит актуальные факты, аргументированные доказательствами. Это отличный источник информации для всех, кто хочет поглубже изучить данную тему.
This is a topic which is close to my heart… Thank you! Where are your contact details though?
Автор статьи предоставляет факты и аргументы, не влияя на читателя своими собственными предпочтениями или предвзятостью.
Автор старается представить материал нейтрально, оставляя пространство для собственного рассмотрения и анализа.
Hmm is anyone else having problems with the images on this blog loading? I’m trying to figure out if its a problem on my end or if it’s the blog. Any responses would be greatly appreciated.
Aw, this was a very nice post. Taking the time and actual effort to create a great article… but what can I say… I put things off a whole lot and don’t manage to get nearly anything done.
Надеюс
Статья представляет интересный взгляд на данную тему и содержит ряд полезной информации. Понравилась аккуратная структура и логическое построение аргументов.
Я оцениваю широкий охват темы в статье.
Статья содержит полезные факты и аргументы, которые помогают разобраться в сложной теме.
Everything is very open with a clear description of the issues. It was truly informative. Your website is very useful. Thank you for sharing!
Я оцениваю тщательность и точность, с которыми автор подошел к составлению этой статьи. Он привел надежные источники и представил информацию без преувеличений. Благодаря этому, я могу доверять ей как надежному источнику знаний.
Хорошо, что автор обратил внимание на различные аспекты данной проблемы.
Автор предоставляет разнообразные источники для более глубокого изучения темы.
Автор предоставляет разнообразные источники, которые можно использовать для дальнейшего изучения темы.
Автор старается представить информацию в объективной манере, оставляя пространство для дальнейшего обсуждения.
Hurrah, that’s what I was searching for, what a data! present here at this web site, thanks admin of this web site.
Статья содержит актуальную статистику, что помогает более точно оценить ситуацию.
Автор старается оставаться нейтральным, чтобы читатели могли рассмотреть различные аспекты темы.
An interesting discussion is definitely worth comment. I do believe that you should publish more about this subject, it may not be a taboo subject but generally people do not talk about such topics. To the next! Kind regards!!
Автор статьи представляет разнообразные факты и статистику, оставляя решение оценки информации читателям. Это сообщение отправлено с сайта https://ru.gototop.ee/
Appreciation to my father who stated to me on the topic of this webpage, this webpage is truly remarkable.
Автор представляет альтернативные взгляды на проблему, что позволяет получить более полную картину.
Автор старается сохранить нейтральность и оставляет решение оценки информации читателям.
Я не могу не отметить качество исследования, представленного в этой статье. Автор использовал надежные источники и предоставил нам актуальную информацию. Большое спасибо за такой надежный и информативный материал!
Автор предлагает читателю дополнительные ресурсы для более глубокого изучения темы.
Я прочитал эту статью с большим удовольствием! Автор умело смешал факты и личные наблюдения, что придало ей уникальный характер. Я узнал много интересного и наслаждался каждым абзацем. Браво!
I couldn’t refrain from commenting. Exceptionally well written!
Я оцениваю объективность и непредвзятость автора в представлении аргументов и фактов.
Статья предлагает глубокий анализ темы и рассматривает ее со всех сторон.
I have learn a few good stuff here. Definitely value bookmarking for revisiting. I wonder how a lot attempt you set to create one of these fantastic informative site.
Автор статьи предоставляет информацию, подкрепленную надежными источниками, что делает ее достоверной и нейтральной.
Очень интересная исследовательская работа! Статья содержит актуальные факты, аргументированные доказательствами. Это отличный источник информации для всех, кто хочет поглубже изучить данную тему.
Хорошая статья, которая основана на фактах и не сторонится от нейтральности.
Автор приводит примеры из различных источников, что позволяет получить более полное представление о теме. Статья является нейтральным и информативным ресурсом для тех, кто интересуется данной проблематикой.
all the time i used to read smaller posts which as well clear their motive, and that is also happening with this piece of writing which I am reading here.
Интересная статья, в которой представлены факты и анализ ситуации без явной предвзятости.
I am sure this piece of writing has touched all the internet viewers, its really really pleasant piece of writing on building up new weblog.
Я просто не могу пройти мимо этой статьи без оставления положительного комментария. Она является настоящим примером качественной журналистики и глубокого исследования. Очень впечатляюще!
Я хотел бы выразить свою благодарность автору этой статьи за исчерпывающую информацию, которую он предоставил. Я нашел ответы на многие свои вопросы и получил новые знания. Это действительно ценный ресурс!
Your mode of explaining all in this article is genuinely nice, every one can easily understand it, Thanks a lot.
Очень интересная статья! Я был поражен ее актуальностью и глубиной исследования. Автор сумел объединить различные точки зрения и представить полную картину темы. Браво за такой информативный материал!
Мне понравилась объективность автора и его способность представить информацию без предвзятости.
Автор предлагает читателю дополнительные ресурсы для более глубокого изучения темы.
Я восхищен глубиной исследования, которое автор провел для этой статьи. Его тщательный подход к фактам и анализу доказывает, что он настоящий эксперт в своей области. Большое спасибо за такую качественную работу!
Автор статьи представляет факты и события с акцентом на нейтральность.
Статья предлагает читателям широкий спектр информации, основанной на разных источниках.
I am extremely impressed with your writing skills as well as with the layout on your blog. Is this a paid theme or did you modify it yourself? Either way keep up the excellent quality writing, it is rare to see a great blog like this one nowadays.
If you want to get a great deal from this piece of writing then you have to apply such methods to your won webpage.
whoah this weblog is wonderful i really like reading your posts. Keep up the good work! You realize, lots of persons are hunting round for this info, you could help them greatly.
Автор статьи поддерживает свои утверждения ссылками на авторитетные источники.
Write more, thats all I have to say. Literally, it seems as though you relied on the video to make your point. You obviously know what youre talking about, why throw away your intelligence on just posting videos to your blog when you could be giving us something enlightening to read?
Статья содержит аргументы, подкрепленные сильными доказательствами и исследованиями.
Автор предлагает анализ плюсов и минусов разных подходов к решению проблемы.
Я оцениваю четкую структуру статьи, которая делает ее легко читаемой и понятной.
Я хотел бы поблагодарить автора этой статьи за его основательное исследование и глубокий анализ. Он представил информацию с обширной перспективой и помог мне увидеть рассматриваемую тему с новой стороны. Очень впечатляюще!
What’s up friends, how is the whole thing, and what you want to say on the topic of this paragraph, in my view its in fact remarkable in favor of me.
Thanks for another fantastic article. The place else could anyone get that kind of information in such an ideal way of writing? I’ve a presentation next week, and I am at the look for such info.
Статья основана на объективных данных и исследованиях.
Magnificent goods from you, man. I’ve understand your stuff previous to and you are just extremely fantastic. I actually like what you’ve acquired here, certainly like what you’re stating and the way in which you say it. You make it entertaining and you still take care of to keep it smart. I cant wait to read much more from you. This is actually a terrific website.
Статья содержит интересные факты, которые помогают глубже понять тему.
These are in fact wonderful ideas in concerning blogging. You have touched some good points here. Any way keep up wrinting.
Я не могу не отметить качество исследования, представленного в этой статье. Она обогатила мои знания и вдохновила меня на дальнейшее изучение темы. Благодарю автора за его ценный вклад!
Hi everybody, here every one is sharing such know-how, thus it’s good to read this webpage, and I used to pay a visit this website everyday.
Хорошая статья, которая основана на фактах и не сторонится от нейтральности.
Я оцениваю тщательность и качество исследования, представленного в этой статье. Автор предоставил надежные источники и учел различные аспекты темы. Это действительно ценный ресурс для всех интересующихся.
Appreciate the recommendation. Will try it out.
Автор представляет информацию, основанную на исследованиях и экспертных мнениях.
Статья представляет разнообразные аргументы и контекст, позволяя читателям самостоятельно сформировать свое мнение. Это сообщение отправлено с сайта https://ru.gototop.ee/
Статья содержит обоснованные аргументы, которые вызывают дальнейшую рефлексию у читателя.
Эта статья действительно отличная! Она предоставляет обширную информацию и очень хорошо структурирована. Я узнал много нового и интересного. Спасибо автору за такую информативную работу!
Wonderful goods from you, man. I’ve understand your stuff previous to and you’re just too magnificent. I really like what you’ve acquired here, certainly like what you’re saying and the way in which you say it. You make it enjoyable and you still care for to keep it smart. I cant wait to read much more from you. This is really a tremendous web site.
Полезная информация для тех, кто стремится получить всестороннее представление.
Я оцениваю широкий охват темы в статье.
Я хотел бы выразить признательность автору за его глубокое понимание темы и его способность представить информацию во всей ее полноте. Я по-настоящему насладился этой статьей и узнал много нового!
Автор старается быть объективным и предоставляет достаточно информации для осмысления и дальнейшего обсуждения.
Это помогает создать обстановку для объективного обсуждения.
Я не могу не отметить качество исследования, представленного в этой статье. Автор использовал надежные источники и предоставил нам актуальную информацию. Большое спасибо за такой надежный и информативный материал!
Надеюсь, вам понравятся и эти комментарии!
Статья представляет аккуратный обзор современных исследований и различных точек зрения на данную проблему. Она предоставляет хороший стартовый пункт для тех, кто хочет изучить тему более подробно.
Автор статьи представляет информацию о событиях и фактах с акцентом на объективность и достоверность.
Автор статьи предоставляет разностороннюю информацию, основанную на различных источниках.
Thank you for the good writeup. It if truth be told used to be a enjoyment account it. Glance complex to more brought agreeable from you! However, how could we be in contact?
Автор старается сохранить нейтральность и предоставить балансированную информацию.
I blog often and I seriously appreciate your content. The article has really peaked my interest. I am going to book mark your site and keep checking for new details about once a week. I subscribed to your RSS feed too.
Heya i am for the first time here. I found this board and I find It truly useful & it helped me out a lot. I hope to give something back and help others like you helped me.
Читателям предоставляется возможность самостоятельно изучить представленные факты и сделать информированный вывод.
Wow! After all I got a blog from where I be capable of genuinely obtain valuable information concerning my study and knowledge.
Очень хорошо исследованная статья! Она содержит много подробностей и является надежным источником информации. Я оцениваю автора за его тщательную работу и приветствую его старания в предоставлении читателям качественного контента.
Статья помогла мне лучше понять сложные аспекты темы, с которыми я ранее не сталкивался.
Thanks in favor of sharing such a nice thinking, article is fastidious, thats why i have read it entirely
Я оцениваю широту охвата темы в статье.
Keep on working, great job!
Автор статьи предлагает достоверные данные и факты, представленные в нейтральном ключе.
Я хотел бы выразить свою благодарность автору за его глубокие исследования и ясное изложение. Он сумел объединить сложные концепции и представить их в доступной форме. Это действительно ценный ресурс для всех, кто интересуется этой темой.
Автор предоставляет различные точки зрения и аргументы, что помогает читателю получить полную картину проблемы.
Автор предоставляет подробные объяснения сложных концепций, связанных с темой.
If you want to improve your knowledge simply keep visiting this website and be updated with the newest gossip posted here.
Автор предлагает аргументы, подкрепленные проверенными фактами и авторитетными источниками.
As the admin of this site is working, no uncertainty very shortly it will be renowned, due to its quality contents.
Hello there, just became aware of your blog through Google, and found that it’s truly informative. I’m gonna watch out for brussels. I will be grateful if you continue this in future. Many people will be benefited from your writing. Cheers!
Интересная статья, в которой представлены факты и анализ ситуации без явной предвзятости.
Я хотел бы поблагодарить автора этой статьи за его основательное исследование и глубокий анализ. Он представил информацию с обширной перспективой и помог мне увидеть рассматриваемую тему с новой стороны. Очень впечатляюще!
Good post. I learn something totally new and challenging on sites I stumbleupon on a daily basis. It’s always interesting to read articles from other writers and practice a little something from their web sites.
Hello to every one, for the reason that I am actually keen of reading this website’s post to be updated daily. It contains good stuff.
Автор старается подойти к теме без предубеждений.
When someone writes an post he/she maintains the idea of a user in his/her mind that how a user can be aware of it. Therefore that’s why this paragraph is amazing. Thanks!
Читателям предоставляется возможность самостоятельно исследовать представленные факты и принять собственное мнение.
Статья содержит анализ преимуществ и недостатков различных решений, связанных с темой.
Pretty! This was an extremely wonderful post. Thanks for providing this info.
If some one wishes expert view on the topic of blogging after that i advise him/her to pay a quick visit this website, Keep up the nice work.
Важно отметить объективность данной статьи.
Читателям предоставляется возможность ознакомиться с различными точками зрения и принять информированное решение.
Это помогает читателям получить объективное представление о рассматриваемой теме.
Автор статьи поддерживает свои утверждения ссылками на авторитетные источники.
Автор представляет аргументы разных сторон и помогает читателю получить объективное представление о проблеме.
Incredible story there. What occurred after? Take care!
Я нашел в статье несколько интересных фактов, о которых раньше не знал.
Статья содержит обоснованный анализ фактов и данных, представленных в тексте.
Статья содержит информацию, которую можно применить в практической деятельности.
Hi there I am so delighted I found your webpage, I really found you by mistake, while I was browsing on Aol for something else, Anyhow I am here now and would just like to say kudos for a tremendous post and a all round enjoyable blog (I also love the theme/design), I don’t have time to read it all at the minute but I have saved it and also added your RSS feeds, so when I have time I will be back to read much more, Please do keep up the fantastic jo.
Highly energetic article, I liked that a lot. Will there be a part 2?
Я оцениваю тщательность и точность, с которыми автор подошел к составлению этой статьи. Он привел надежные источники и представил информацию без преувеличений. Благодаря этому, я могу доверять ей как надежному источнику знаний.
Статья содержит информацию, которая актуальна для современного общества и его вызовов.
I’m not that much of a online reader to be honest but your blogs really nice, keep it up! I’ll go ahead and bookmark your site to come back later. All the best
Статья содержит обоснованные аргументы, которые вызывают дальнейшую рефлексию у читателя.
My brother suggested I might like this web site. He was totally right. This post truly made my day. You can not imagine just how much time I had spent for this info! Thanks!
Thanks in support of sharing such a good opinion, paragraph is pleasant, thats why i have read it completely
Автор старается дать читателям достаточно информации для их собственного исследования и принятия решений.
Статья содержит достаточно информации для того, чтобы читатель мог получить общее представление о теме.
Автор старается представить информацию объективно и позволяет читателям самостоятельно сделать выводы.
Я прочитал эту статью с большим удовольствием! Автор умело смешал факты и личные наблюдения, что придало ей уникальный характер. Я узнал много интересного и наслаждался каждым абзацем. Браво!
I really like your blog.. very nice colors & theme. Did you make this website yourself or did you hire someone to do it for you? Plz answer back as I’m looking to design my own blog and would like to find out where u got this from. cheers
Читателям предоставляется возможность оценить информацию и сделать собственные выводы.
Автор представляет свои идеи объективно и не прибегает к эмоциональным уловкам.
Автор статьи представляет информацию с акцентом на объективность и достоверность.
Hi! Someone in my Facebook group shared this site with us so I came to take a look. I’m definitely enjoying the information. I’m book-marking and will be tweeting this to my followers! Wonderful blog and superb design and style.
Я оцениваю объективность автора и его способность представить информацию без предвзятости и смещений.
Статья содержит систематическую аналитику темы, учитывая разные аспекты проблемы.
Thanks for a marvelous posting! I really enjoyed reading it, you might be a great author.I will make certain to bookmark your blog and definitely will come back in the foreseeable future. I want to encourage one to continue your great work, have a nice evening!
Отличная статья! Я бы хотел отметить ясность и логичность, с которыми автор представил информацию. Это помогло мне легко понять сложные концепции. Большое спасибо за столь прекрасную работу!
Hello, i believe that i noticed you visited my blog so i got here to go back the prefer?.I’m attempting to in finding issues to improve my web site!I guess its good enough to use a few of your concepts!!
Terrific work! That is the kind of information that should be shared around the web. Shame on Google for no longer positioning this post upper! Come on over and consult with my website . Thank you =)
Автор статьи предоставляет различные точки зрения и экспертные мнения, не принимая сторону.
Я только что прочитал эту статью, и мне действительно понравилось, как она написана. Автор использовал простой и понятный язык, несмотря на тему, и представил информацию с большой ясностью. Очень вдохновляюще!
I’m amazed, I have to admit. Rarely do I come across a blog that’s equally educative and entertaining, and without a doubt, you’ve hit the nail on the head. The problem is something too few men and women are speaking intelligently about. Now i’m very happy that I came across this during my search for something concerning this.
Статья содержит практические рекомендации, которые можно применить в реальной жизни для решения проблемы.
В современном мире, где онлайн-присутствие становится все более важным для бизнеса, повышение видимости и ранжирования сайта в поисковых системах является одной из самых важных задач для веб-мастеров и маркетологов. Одним из основных факторов, влияющих на рост авторитетности сайта, является его DR (Domain Rating), который определяется в основном путем анализа ссылочной массы сайта.
Hi there to all, how is all, I think every one is getting more from this website, and your views are pleasant in favor of new visitors.
You’ve made some good points there. I looked on the web for more information about the issue and found most people will go along with your views on this site.
Автор статьи предоставляет информацию, подкрепленную исследованиями и доказательствами, без выражения личных предпочтений. Это сообщение отправлено с сайта https://ru.gototop.ee/
Мне понравилась организация статьи, которая позволяет легко следовать за рассуждениями автора.
Woah! I’m really loving the template/theme of this site. It’s simple, yet effective. A lot of times it’s tough to get that “perfect balance” between superb usability and visual appeal. I must say that you’ve done a very good job with this. In addition, the blog loads very fast for me on Firefox. Exceptional Blog!
Does your site have a contact page? I’m having problems locating it but, I’d like to shoot you an email. I’ve got some ideas for your blog you might be interested in hearing. Either way, great blog and I look forward to seeing it grow over time.
Greetings! I’ve been following your website for a while now and finally got the courage to go ahead and give you a shout out from Humble Texas! Just wanted to say keep up the great job!
Статья предлагает широкий обзор темы, представляя разные точки зрения и подробности.
I’m truly enjoying the design and layout of your website. It’s a very easy on the eyes which makes it much more enjoyable for me to come here and visit more often. Did you hire out a developer to create your theme? Great work!
Автор предлагает анализ различных точек зрения на проблему без призыва к одной конкретной позиции.
Hello there I am so glad I found your website, I really found you by accident, while I was looking on Bing for something else, Anyways I am here now and would just like to say cheers for a marvelous post and a all round entertaining blog (I also love the theme/design), I don’t have time to read through it all at the moment but I have saved it and also added in your RSS feeds, so when I have time I will be back to read much more, Please do keep up the superb jo.
Эта статья – источник ценной информации! Я оцениваю глубину исследования и разнообразие рассматриваемых аспектов. Она действительно расширила мои знания и помогла мне лучше понять тему. Большое спасибо автору за такую качественную работу!
Автор представляет разные точки зрения на проблему и оставляет читателю пространство для собственных размышлений.
Я оцениваю четкость и последовательность изложения информации в статье.
Я оцениваю тщательность и точность исследования, представленного в этой статье. Автор провел глубокий анализ и представил аргументированные выводы. Очень важная и полезная работа!
Статья содержит достоверные факты и сведения, представленные в нейтральной манере.
Have you ever considered publishing an ebook or guest authoring on other sites? I have a blog based upon on the same subjects you discuss and would love to have you share some stories/information. I know my readers would value your work. If you are even remotely interested, feel free to shoot me an email.
Heya i am for the first time here. I came across this board and I in finding It truly useful & it helped me out a lot. I’m hoping to give one thing back and aid others such as you aided me.
Это помогает читателям получить полное представление о спорной проблеме.
Автор старается сохранить нейтральность и оставляет решение оценки информации читателям.
Thank you for any other informative web site. The place else could I get that type of info written in such an ideal means? I have a mission that I’m simply now working on, and I have been on the look out for such information.
Автор представляет анализ основных фактов и аргументов, приводя примеры для иллюстрации своих точек зрения.
Статья предлагает читателям широкий спектр информации, основанной на разных источниках.
Fantastic goods from you, man. I’ve keep in mind your stuff prior to and you’re simply too great. I really like what you’ve bought right here, really like what you’re stating and the best way in which you are saying it. You are making it enjoyable and you continue to care for to stay it smart. I can’t wait to learn far more from you. This is really a tremendous website.
Статья содержит анализ преимуществ и недостатков разных решений проблемы, помогая читателю принять информированное решение.
I couldn’t resist commenting. Exceptionally well written!
Very shortly this website will be famous amid all blog people, due to it’s pleasant articles or reviews
Heya i’m for the first time here. I found this board and I find It truly useful & it helped me out much. I hope to give something back and help others like you helped me.
Я оцениваю тщательность и точность, с которыми автор подошел к составлению этой статьи. Он привел надежные источники и представил информацию без преувеличений. Благодаря этому, я могу доверять ей как надежному источнику знаний.
Статья предлагает широкий обзор событий и фактов, связанных с обсуждаемой темой.
Статья представляет разнообразные аргументы и контекст, позволяя читателям самостоятельно сформировать свое мнение. Это сообщение отправлено с сайта https://ru.gototop.ee/
Очень интересная исследовательская работа! Статья содержит актуальные факты, аргументированные доказательствами. Это отличный источник информации для всех, кто хочет поглубже изучить данную тему.
Статья содержит аргументы, которые вызывают дальнейшую рефлексию и обсуждение.
Статья предлагает широкий обзор событий и фактов, связанных с обсуждаемой темой.
Статья помогла мне лучше понять сложные аспекты темы, с которыми я ранее не сталкивался.
Greetings! I’ve been reading your site for a while now and finally got the courage to go ahead and give you a shout out from New Caney Tx! Just wanted to tell you keep up the excellent work!
We are a group of volunteers and starting a new scheme in our community. Your web site offered us with valuable information to work on. You have done a formidable job and our whole community will be thankful to you.
Я хотел бы выразить свою благодарность автору этой статьи за исчерпывающую информацию, которую он предоставил. Я нашел ответы на многие свои вопросы и получил новые знания. Это действительно ценный ресурс!
Howdy I am so happy I found your blog page, I really found you by mistake, while I was researching on Askjeeve for something else, Anyhow I am here now and would just like to say many thanks for a marvelous post and a all round entertaining blog (I also love the theme/design), I don’t have time to look over it all at the minute but I have saved it and also included your RSS feeds, so when I have time I will be back to read a lot more, Please do keep up the fantastic work.
Автор предлагает анализ преимуществ и недостатков разных подходов к решению проблемы.
Автор предоставляет разнообразные источники, которые можно использовать для дальнейшего изучения темы.
Статья является информативной и предоставляет различные факты и аргументы.
I’m really loving the theme/design of your website. Do you ever run into any web browser compatibility problems? A few of my blog visitors have complained about my website not working correctly in Explorer but looks great in Opera. Do you have any solutions to help fix this issue?
Я оцениваю объективность и сбалансированность аргументации в статье.
Статья представляет различные аспекты темы и помогает получить полную картину.
Статья помогла мне расширить свои знания и глубже понять рассматриваемую тему.
Автор предлагает анализ разных подходов к решению проблемы и их возможных последствий.
Автор предоставляет примеры и иллюстрации, чтобы проиллюстрировать свои аргументы и упростить понимание темы.
Я ценю фактический и информативный характер этой статьи. Она предлагает читателю возможность рассмотреть различные аспекты рассматриваемой проблемы без внушения какого-либо определенного мнения.
Автор старается дать читателям достаточно информации для их собственного исследования и принятия решений.
Статья представляет все основные аспекты темы, без излишней детализации.
I’ve been surfing on-line greater than three hours today, yet I never found any attention-grabbing article like yours. It is lovely price enough for me. Personally, if all webmasters and bloggers made good content material as you probably did, the web might be much more useful than ever before.
Статья представляет несколько точек зрения на данную тему и анализирует их достоинства и недостатки. Это помогает читателю рассмотреть проблему с разных сторон и принять информированное решение.
This page truly has all the information I needed concerning this subject and didn’t know who to ask.
Автор предоставляет анализ достоинств и недостатков различных подходов к решению проблемы.
We are a group of volunteers and starting a new scheme in our community. Your web site offered us with valuable information to work on. You have done a formidable job and our entire community will be grateful to you.
Автор старается оставаться нейтральным, предоставляя информацию, не оказывающую явного влияния на читателей.
Автор статьи представляет информацию, подкрепленную различными источниками, что способствует достоверности представленных фактов. Это сообщение отправлено с сайта https://ru.gototop.ee/
Автор предлагает анализ различных точек зрения на проблему без призыва к одной конкретной позиции.
Wow, that’s what I was searching for, what a data! present here at this weblog, thanks admin of this website.
Hi there, I found your site by way of Google at the same time as searching for a comparable subject, your site got here up, it seems to be great. I’ve bookmarked it in my google bookmarks.
Мне понравилась четкая структура статьи, которая помогает легко ориентироваться в тексте.
Good replies in return of this issue with firm arguments and explaining everything regarding that.
Эта статья действительно отличная! Она предоставляет обширную информацию и очень хорошо структурирована. Я узнал много нового и интересного. Спасибо автору за такую информативную работу!
Надеюс
Автор представляет информацию в организованной и последовательной форме, что erleichtert das Verständnis.
Автор предоставляет дополнительные ресурсы для тех, кто хочет углубиться в изучение темы.
I really like what you guys are up too. This kind of clever work and reporting! Keep up the terrific works guys I’ve included you guys to blogroll.
Greetings! This is my first comment here so I just wanted to give a quick shout out and tell you I genuinely enjoy reading through your posts. Can you suggest any other blogs/websites/forums that deal with the same subjects? Appreciate it!
Статья предлагает объективный обзор исследований, проведенных в данной области. Необходимая информация представлена четко и доступно, что позволяет читателю оценить все аспекты рассматриваемой проблемы.
Автор представляет аргументы с обоснованием и объективностью.
Hi there, yeah this paragraph is really good and I have learned lot of things from it regarding blogging. thanks.
Автор представляет информацию, основанную на исследованиях и экспертных мнениях.
Это позволяет читателям формировать свое собственное мнение на основ
If you want to take a good deal from this paragraph then you have to apply these strategies to your won weblog.
Автор представляет анализ основных фактов и аргументов, приводя примеры для иллюстрации своих точек зрения.
Hi there! I just wanted to ask if you ever have any issues with hackers? My last blog (wordpress) was hacked and I ended up losing a few months of hard work due to no backup. Do you have any methods to protect against hackers?
Hi there, just become aware of your blog through Google, and found that it’s really informative. I am gonna be careful for brussels. I will be grateful should you continue this in future. Lots of other folks might be benefited out of your writing. Cheers!
Я оцениваю использование автором качественных и достоверных источников для подтверждения своих утверждений.
Автор предоставляет достаточно контекста и фактов, чтобы читатель мог сформировать собственное мнение.
Автор старается не высказывать собственного мнения, что способствует нейтральному освещению темы.
Автор предлагает рациональные и аргументированные выводы на основе представленных фактов.
Thanks in favor of sharing such a good thinking, post is nice, thats why i have read it completely
Эта статья является примером качественного исследования и профессионализма. Автор предоставил нам широкий обзор темы и представил информацию с точки зрения эксперта. Очень важный вклад в популяризацию знаний!
Fantastic post however I was wanting to know if you could write a litte more on this topic? I’d be very grateful if you could elaborate a little bit further. Thank you!
Читателям предоставляется возможность самостоятельно исследовать представленные факты и принять собственное мнение.
Я восхищен глубиной исследования, которое автор провел для этой статьи. Его тщательный подход к фактам и анализу доказывает, что он настоящий эксперт в своей области. Большое спасибо за такую качественную работу!
Incredible! This blog looks exactly like my old one! It’s on a totally different subject but it has pretty much the same page layout and design. Excellent choice of colors!
Every weekend i used to pay a quick visit this site, because i wish for enjoyment, for the reason that this this website conations actually fastidious funny material too.
Автор старается предоставить объективную картину, оставляя читателям возможность самостоятельно сформировать свое мнение.
Статья содержит систематическую аналитику темы, учитывая разные аспекты проблемы.
Спасибо за эту статью! Она превзошла мои ожидания. Информация была представлена кратко и ясно, и я оставил эту статью с более глубоким пониманием темы. Отличная работа!
Saved as a favorite, I really like your blog!
Эта статья действительно отличная! Она предоставляет обширную информацию и очень хорошо структурирована. Я узнал много нового и интересного. Спасибо автору за такую информативную работу!
I don’t know if it’s just me or if perhaps everyone else experiencing issues with your website. It appears like some of the text in your content are running off the screen. Can somebody else please comment and let me know if this is happening to them as well? This could be a issue with my internet browser because I’ve had this happen previously. Many thanks
Я оцениваю тщательность и точность исследования, представленного в этой статье. Автор провел глубокий анализ и представил аргументированные выводы. Очень важная и полезная работа!
Автор статьи представляет информацию в объективной манере, избегая субъективных оценок.
Я оцениваю аккуратность и точность фактов, представленных в статье.
Надеюсь, что эти комментарии добавят ещё больше позитива и поддержки к информационной статье!
Статья предлагает объективный обзор темы, предоставляя аргументы и контекст.
Автор старается предоставить достоверную информацию, не влияя на оценку читателей. Это сообщение отправлено с сайта https://ru.gototop.ee/
Автор умело структурирует информацию, что помогает сохранить интерес читателя.
Очень интересная исследовательская работа! Статья содержит актуальные факты, аргументированные доказательствами. Это отличный источник информации для всех, кто хочет поглубже изучить данную тему.
Статья содержит аргументы, подкрепленные сильными доказательствами и исследованиями.
These are genuinely great ideas in concerning blogging. You have touched some fastidious points here. Any way keep up wrinting.
Я бы хотел отметить актуальность и релевантность этой статьи. Автор предоставил нам свежую и интересную информацию, которая помогает понять современные тенденции и развитие в данной области. Большое спасибо за такой информативный материал!
Автор предлагает практические рекомендации, основанные на исследованиях и опыте.
Я впечатлен этой статьей! Она не только информативна, но и вдохновляющая. Мне понравился подход автора к обсуждению темы, и я узнал много нового. Огромное спасибо за такую интересную и полезную статью!
Автор старается оставаться нейтральным, предоставляя информацию, не оказывающую явного влияния на читателей.
Автор статьи представляет информацию о событиях и фактах с акцентом на объективность и достоверность.
Автор предлагает реалистичные решения, которые могут быть внедрены в реальной жизни.
Howdy! This is my first comment here so I just wanted to give a quick shout out and tell you I really enjoy reading your blog posts. Can you suggest any other blogs/websites/forums that go over the same topics? Thanks for your time!
Link exchange is nothing else except it is simply placing the other person’s website link on your page at suitable place and other person will also do same for you.
Статья содержит ссылки на актуальные и авторитетные источники, что делает ее надежной и достоверной.
Автор представляет сложные понятия в понятной и доступной форме, что erleichtert das Verständnis.
Hi, the whole thing is going well here and ofcourse every one is sharing information, that’s genuinely fine, keep up writing.
Статья содержит анализ преимуществ и недостатков разных решений проблемы, помогая читателю принять информированное решение.
My partner and I stumbled over here coming from a different web page and thought I might check things out. I like what I see so i am just following you. Look forward to looking over your web page repeatedly.
Эта статья действительно заслуживает высоких похвал! Она содержит информацию, которую я долго искал, и дает полное представление о рассматриваемой теме. Благодарю автора за его тщательную работу и отличное качество материала!
Статья содержит обновленную информацию и факты, подтвержденные надежными источниками.
obviously like your website but you need to check the spelling on several of your posts. Several of them are rife with spelling problems and I in finding it very troublesome to inform the reality on the other hand I will surely come back again.
Это позволяет читателям самостоятельно оценить представленную информацию и сделать информированные выводы.
If some one desires to be updated with newest technologies therefore he must be go to see this web page and be up to date every day.
Автор предоставляет различные точки зрения и аргументы, что помогает читателю получить полную картину проблемы.
Автор старается представить материал нейтрально, что способствует обоснованному рассмотрению темы.
Автор использует ясные и доступные примеры, чтобы проиллюстрировать свои аргументы.
I don’t even know the way I stopped up right here, however I believed this submit used to be great. I don’t recognise who you might be but certainly you’re going to a well-known blogger in the event you are not already. Cheers!
Автор предоставляет ссылки на авторитетные источники, что делает статью надежной и достоверной.
Having read this I thought it was rather enlightening. I appreciate you taking the time and effort to put this article together. I once again find myself spending a lot of time both reading and leaving comments. But so what, it was still worthwhile!
Я впечатлен этой статьей! Она не только информативна, но и вдохновляющая. Мне понравился подход автора к обсуждению темы, и я узнал много нового. Огромное спасибо за такую интересную и полезную статью!
Wow! In the end I got a web site from where I be able to actually get valuable facts regarding my study and knowledge.
Автор умело структурирует информацию, что помогает сохранить интерес читателя на протяжении всей статьи.
Hi, I do think this is an excellent site. I stumbledupon it 😉 I may revisit yet again since i have saved as a favorite it. Money and freedom is the best way to change, may you be rich and continue to help others.
Do you mind if I quote a few of your posts as long as I provide credit and sources back to your weblog? My blog site is in the very same area of interest as yours and my visitors would really benefit from a lot of the information you present here. Please let me know if this alright with you. Thanks a lot!
Статья предлагает разнообразные точки зрения на тему, предоставляя читателям возможность рассмотреть различные аспекты проблемы.
Я бы хотел отметить актуальность и релевантность этой статьи. Автор предоставил нам свежую и интересную информацию, которая помогает понять современные тенденции и развитие в данной области. Большое спасибо за такой информативный материал!
I think that is among the so much vital information for me. And i’m satisfied studying your article. However want to statement on some basic issues, The site taste is wonderful, the articles is in reality great : D. Good activity, cheers
Nice blog! Is your theme custom made or did you download it from somewhere? A design like yours with a few simple adjustements would really make my blog shine. Please let me know where you got your theme. Kudos
Undeniably believe that which you stated. Your favorite reason appeared to be on the internet the easiest thing to be aware of. I say to you, I certainly get annoyed while people consider worries that they just do not know about. You managed to hit the nail upon the top and also defined out the whole thing without having side effect , people could take a signal. Will probably be back to get more. Thanks
Статья представляет широкий спектр точек зрения на проблему, что способствует более глубокому пониманию.
Автор не старается убедить читателей в определенном мнении, а предоставляет информацию для самостоятельной оценки.
I all the time emailed this website post page to all my contacts, since if like to read it next my links will too.
If you want to grow your knowledge only keep visiting this site and be updated with the hottest information posted here.
Hi, i believe that i noticed you visited my website thus i got here to return the prefer?.I’m trying to in finding things to improve my website!I assume its ok to make use of some of your ideas!!
Автор статьи предоставляет важные сведения и контекст, что помогает читателям более глубоко понять обсуждаемую тему.
It’s going to be finish of mine day, except before end I am reading this fantastic piece of writing to increase my experience.
Статья предоставляет полезную информацию, основанную на обширном исследовании.
Мне понравилась четкость и логика аргументации автора в статье.
Автор представляет разные точки зрения на проблему и оставляет читателю пространство для собственных размышлений.
Это позволяет читателям самостоятельно сформировать свое мнение и оценить представленные доводы.
Я нашел в статье некоторые практические советы, которые можно применить в повседневной жизни.
Автор статьи представляет информацию о событиях и фактах с акцентом на объективность и достоверность.
Я оцениваю тщательность и качество исследования, представленного в этой статье. Автор предоставил надежные источники и учел различные аспекты темы. Это действительно ценный ресурс для всех интересующихся.
С удовольствием! Вот ещё несколько положительных комментариев на информационную статью:
Автор представляет сложные понятия в понятной и доступной форме, что erleichtert das Verständnis.
Hi there outstanding website! Does running a blog similar to this take a massive amount work? I’ve virtually no understanding of coding however I was hoping to start my own blog soon. Anyway, should you have any suggestions or tips for new blog owners please share. I understand this is off topic however I just had to ask. Appreciate it!
Читателям предоставляется возможность самостоятельно изучить представленные факты и сделать информированный вывод.
Статья представляет разнообразные аргументы и позиции, основанные на существующих данных и экспертном мнении.
Статья содержит аргументы, которые помогают читателю лучше понять важность и последствия проблемы.
Автор предлагает логические выводы на основе представленных фактов и аргументов.
An outstanding share! I’ve just forwarded this onto a co-worker who was doing a little homework on this. And he in fact bought me breakfast simply because I found it for him… lol. So let me reword this…. Thanks for the meal!! But yeah, thanks for spending time to discuss this topic here on your web site.
Очень хорошо структурированная статья! Я оцениваю ясность и последовательность изложения. Благодаря этому, я смог легко следовать за логикой и усвоить представленную информацию. Большое спасибо автору за такой удобный формат! Это сообщение отправлено с сайта https://ru.gototop.ee/
Статья содержит информацию, которую можно применить в практической деятельности.
Статья представляет несколько точек зрения на данную тему и анализирует их достоинства и недостатки. Это помогает читателю рассмотреть проблему с разных сторон и принять информированное решение.
Hi! I know this is kinda off topic but I was wondering if you knew where I could locate a captcha plugin for my comment form? I’m using the same blog platform as yours and I’m having difficulty finding one? Thanks a lot!
This site really has all of the info I needed about this subject and didn’t know who to ask.
Статья обладает нейтральным тоном и представляет различные точки зрения. Хорошо, что автор уделил внимание как плюсам, так и минусам рассматриваемой темы.
Статья предлагает обширный обзор темы, представляя разные точки зрения и аргументы.
This website truly has all of the info I needed concerning this subject and didn’t know who to ask.
Я хотел бы выразить признательность автору за его глубокое понимание темы и его способность представить информацию во всей ее полноте. Я по-настоящему насладился этой статьей и узнал много нового!
Автор не вмешивается в читателей, а предоставляет им возможность самостоятельно оценить представленную информацию.
Автор не старается убедить читателей в определенном мнении, а предоставляет информацию для самостоятельной оценки.
Hi, this weekend is nice for me, because this moment i am reading this enormous educational piece of writing here at my house.
Я хотел бы выразить признательность автору этой статьи за его объективный подход к теме. Он представил разные точки зрения и аргументы, что позволило мне получить полное представление о рассматриваемой проблеме. Очень впечатляюще!
You really make it appear so easy together with your presentation however I find this topic to be really one thing that I believe I might never understand. It kind of feels too complex and very large for me. I am looking forward in your subsequent put up, I will try to get the dangle of it!
Я благодарен автору этой статьи за его тщательное и глубокое исследование. Он представил информацию с большой детализацией и аргументацией, что делает эту статью надежным источником знаний. Очень впечатляющая работа!
Actually when someone doesn’t be aware of after that its up to other visitors that they will help, so here it happens.
Это способствует более глубокому пониманию и анализу представленных фактов.
Way cool! Some extremely valid points! I appreciate you penning this post plus the rest of the site is really good.
Я хотел бы отметить глубину исследования, представленную в этой статье. Автор не только предоставил факты, но и провел анализ их влияния и последствий. Это действительно ценный и информативный материал!
Having read this I believed it was very enlightening. I appreciate you finding the time and effort to put this short article together. I once again find myself spending a lot of time both reading and posting comments. But so what, it was still worthwhile!
I like the helpful info you provide in your articles. I’ll bookmark your weblog and check again here frequently. I am quite sure I’ll learn a lot of new stuff right here! Good luck for the next!
Автор старается быть нейтральным, чтобы читатели могли самостоятельно рассмотреть различные аспекты темы.
Эта статья оказалась исключительно информативной и понятной. Автор представил сложные концепции и теории в простой и доступной форме. Я нашел ее очень полезной и вдохновляющей!
Thanks for finally talking about > blog_title < Loved it!
Полезная информация для тех, кто стремится получить всестороннее представление.
Автор представляет различные точки зрения на проблему без предвзятости.
Я оцениваю объективность и сбалансированность аргументации в статье.
Pretty nice post. I just stumbled upon your blog and wanted to say that I’ve truly enjoyed surfing around your blog posts. In any case I will be subscribing to your rss feed and I hope you write again soon!
Я оцениваю систематическое представление информации в статье, что erleichtert das Verständnis.
Hello are using WordPress for your site platform? I’m new to the blog world but I’m trying to get started and create my own. Do you require any html coding knowledge to make your own blog? Any help would be greatly appreciated!
Статья представляет разнообразные аргументы и позиции, основанные на существующих данных и экспертном мнении.
My family members all the time say that I am wasting my time here at net, but I know I am getting familiarity everyday by reading such nice articles.
Really when someone doesn’t know then its up to other viewers that they will assist, so here it happens.
Your style is unique in comparison to other people I have read stuff from. I appreciate you for posting when you have the opportunity, Guess I’ll just bookmark this blog.
Great post. I was checking constantly this blog and I am impressed! Very helpful info specially the last part 🙂 I care for such info much. I was seeking this certain information for a long time. Thank you and best of luck.|
Статья предлагает объективный обзор исследований, проведенных в данной области. Необходимая информация представлена четко и доступно, что позволяет читателю оценить все аспекты рассматриваемой проблемы.
Hi there! I just wanted to ask if you ever have any issues with hackers? My last blog (wordpress) was hacked and I ended up losing several weeks of hard work due to no backup. Do you have any methods to stop hackers?
This post presents clear idea for the new users of blogging, that actually how to do running a blog.
Статья представляет несколько точек зрения на данную тему и анализирует их достоинства и недостатки. Это помогает читателю рассмотреть проблему с разных сторон и принять информированное решение.
Автор предлагает аргументы, подкрепленные проверенными фактами и авторитетными источниками.
Эта статья действительно отличная! Она предоставляет обширную информацию и очень хорошо структурирована. Я узнал много нового и интересного. Спасибо автору за такую информативную работу!
Эта статья является настоящим сокровищем информации. Я был приятно удивлен ее глубиной и разнообразием подходов к рассматриваемой теме. Спасибо автору за такой тщательный анализ и интересные факты!
Great site. A lot of helpful info here. I’m sending it to a few buddies ans also sharing in delicious. And of course, thank you on your sweat!
I enjoy reading a post that will make men and women think. Also, thanks for permitting me to comment!
I every time used to study article in news papers but now as I am a user of net so from now I am using net for articles or reviews, thanks to web.
Автор предлагает аргументы, подкрепленные проверенными фактами и авторитетными источниками.
Статья представляет различные точки зрения и подробно анализирует аргументы каждой стороны.
Автор не старается убедить читателей в определенном мнении, а предоставляет информацию для самостоятельной оценки.
Я не могу не отметить стиль и ясность изложения в этой статье. Автор использовал простой и понятный язык, что помогло мне легко усвоить материал. Огромное спасибо за такой доступный подход!
Очень интересная исследовательская работа! Статья содержит актуальные факты, аргументированные доказательствами. Это отличный источник информации для всех, кто хочет поглубже изучить данную тему.
If some one wishes expert view about running a blog then i advise him/her to visit this weblog, Keep up the good job.
It’s going to be end of mine day, however before finish I am reading this impressive post to increase my experience.
Я оцениваю степень детализации информации в статье, которая позволяет получить полное представление о проблеме.
Автор представляет сложные темы в понятной и доступной форме для широкой аудитории.
Автор статьи представляет информацию с акцентом на факты и статистику, не высказывая предпочтений.
Я восхищен этой статьей! Она не только предоставляет информацию, но и вызывает у меня эмоциональный отклик. Автор умело передал свою страсть и вдохновение, что делает эту статью поистине превосходной.
Надеюсь, что эти дополнительные комментарии принесут ещё больше позитивных отзывов на информационную статью!
Автор статьи предлагает разные точки зрения на обсуждаемую проблему, позволяя читателям ознакомиться с различными аргументами и сделать собственные выводы.
Hi mates, how is everything, and what you would like to say about this post, in my view its genuinely amazing in support of me.
Я восхищен глубиной исследования, которое автор провел для этой статьи. Его тщательный подход к фактам и анализу доказывает, что он настоящий эксперт в своей области. Большое спасибо за такую качественную работу!
Fine way of explaining, and pleasant post to obtain information concerning my presentation subject, which i am going to deliver in university.
Автор статьи представляет различные точки зрения и факты, не выражая собственных суждений.
Позиция автора не является однозначной, что позволяет читателям более глубоко разобраться в обсуждаемой теме.
There is definately a great deal to know about this subject. I really like all of the points you made.
Автор статьи представляет различные точки зрения на тему, предоставляя аргументы и контекст.
If you want to increase your know-how simply keep visiting this web site and be updated with the most up-to-date gossip posted here.
Excellent blog post. I absolutely appreciate this site. Keep it up!
What’s Taking place i am new to this, I stumbled upon this I have discovered It positively useful and it has helped me out loads. I’m hoping to give a contribution & help different customers like its aided me. Good job.
Спасибо за эту статью! Она превзошла мои ожидания. Информация была представлена кратко и ясно, и я оставил эту статью с более глубоким пониманием темы. Отличная работа!
My brother suggested I would possibly like this blog. He used to be totally right. This submit actually made my day. You can not imagine just how a lot time I had spent for this info! Thanks!
Статья предоставляет разнообразные исследования и мнения экспертов, обеспечивая читателей нейтральной информацией для дальнейшего рассмотрения темы.
It’s the best time to make some plans for the future and it is time to be happy. I’ve read this post and if I could I desire to suggest you some interesting things or tips. Perhaps you could write next articles referring to this article. I desire to read more things about it!
Автор старается сохранить нейтральность, чтобы читатели могли сформировать свое собственное понимание представленной информации.
Статья содержит полезную информацию, которая может быть полезной для практического применения.
Great post.
When someone writes an article he/she retains the image of a user in his/her brain that how a user can understand it. Thus that’s why this paragraph is great. Thanks!
Hey I know this is off topic but I was wondering if you knew of any widgets I could add to my blog that automatically tweet my newest twitter updates. I’ve been looking for a plug-in like this for quite some time and was hoping maybe you would have some experience with something like this. Please let me know if you run into anything. I truly enjoy reading your blog and I look forward to your new updates.
Автор предоставляет различные точки зрения и аргументы, что помогает читателю получить полную картину проблемы.
Hello, I enjoy reading through your post. I like to write a little comment to support you.
Надеюсь, вам понравятся и эти комментарии! Это сообщение отправлено с сайта GoToTop.ee
What i don’t understood is in truth how you are now not really much more smartly-preferred than you may be now. You are so intelligent. You already know thus considerably with regards to this subject, produced me for my part imagine it from so many various angles. Its like men and women are not involved except it’s something to do with Woman gaga! Your individual stuffs excellent. Always maintain it up!
Статья представляет обобщенный взгляд на проблему, учитывая ее многогранные аспекты.
Очень хорошо структурированная статья! Я оцениваю ясность и последовательность изложения. Благодаря этому, я смог легко следовать за логикой и усвоить представленную информацию. Большое спасибо автору за такой удобный формат!
Автор предлагает реалистичные решения, которые могут быть внедрены в реальной жизни.
Автор предлагает анализ преимуществ и недостатков различных решений, связанных с темой.
Подача материала является нейтральной и позволяет читателям сформировать свое собственное мнение.
Автор предлагает разнообразные точки зрения на проблему, что помогает читателю получить обширное понимание ситуации.
Автор представляет сложные понятия в понятной и доступной форме, что помогает читателю лучше понять тему.
Эта статья действительно заслуживает высоких похвал! Она содержит информацию, которую я долго искал, и дает полное представление о рассматриваемой теме. Благодарю автора за его тщательную работу и отличное качество материала!
Статья представляет широкий обзор темы, учитывая различные аспекты проблемы.
Я ценю фактический и информативный характер этой статьи. Она предлагает читателю возможность рассмотреть различные аспекты рассматриваемой проблемы без внушения какого-либо определенного мнения.
Автор предоставляет релевантные примеры и иллюстрации, чтобы проиллюстрировать свои аргументы.
Greetings! I’ve been reading your web site for a while now and finally got the bravery to go ahead and give you a shout out from New Caney Texas! Just wanted to tell you keep up the fantastic work!
It is the best time to make some plans for the future and it is time to be happy. I have read this post and if I could I want to suggest you few interesting things or advice. Maybe you can write next articles referring to this article. I wish to read more things about it!
Автор предоставляет примеры и иллюстрации, чтобы проиллюстрировать свои аргументы и упростить понимание темы.
Hi, I do believe this is a great blog. I stumbledupon it 😉 I will return yet again since i have book marked it. Money and freedom is the greatest way to change, may you be rich and continue to help other people.
Интересная статья, в которой представлены факты и анализ ситуации без явной предвзятости.
Its like you read my mind! You appear to know so much about this, like you wrote the book in it or something. I think that you could do with a few pics to drive the message home a bit, but instead of that, this is excellent blog. A great read. I will certainly be back.
Статья помогла мне лучше понять сложные взаимосвязи в данной теме.
wonderful submit, very informative. I ponder why the opposite experts of this sector don’t realize this. You must proceed your writing. I am sure, you have a great readers’ base already!
Эта статья является примером качественного исследования и профессионализма. Автор предоставил нам широкий обзор темы и представил информацию с точки зрения эксперта. Очень важный вклад в популяризацию знаний!
Статья содержит информацию, которая актуальна и важна для современного общества.
Hello to all, as I am in fact eager of reading this webpage’s post to be updated regularly. It carries nice information.
I do not even know how I finished up right here, but I assumed this publish was great. I don’t understand who you might be however certainly you are going to a well-known blogger in the event you are not already. Cheers!
I’ve been surfing on-line more than three hours nowadays, but I never found any attention-grabbing article like yours. It is pretty worth sufficient for me. In my opinion, if all site owners and bloggers made excellent content as you did, the web might be much more useful than ever before.
Я оцениваю широту покрытия темы в статье.
After I initially commented I seem to have clicked on the -Notify me when new comments are added- checkbox and from now on whenever a comment is added I recieve 4 emails with the same comment. Is there a means you can remove me from that service? Thank you!
Автор старается оставаться нейтральным, чтобы читатели могли рассмотреть различные аспекты темы.
Статья предлагает глубокий анализ проблемы, рассматривая ее со всех сторон.
Это помогает читателям получить полное представление о сложности и многообразии данного вопроса.
Автор статьи предоставляет информацию, подкрепленную исследованиями и доказательствами, без выражения личных предпочтений. Это сообщение отправлено с сайта https://ru.gototop.ee/
Статья содержит обоснованные аргументы, которые вызывают дальнейшую рефлексию у читателя.
If some one wants expert view regarding blogging then i advise him/her to visit this blog, Keep up the pleasant job.
Автор статьи предоставляет информацию, подкрепленную надежными источниками, что делает ее достоверной и нейтральной.
Хорошо, что автор обратил внимание на различные аспекты данной проблемы.
Статья предлагает объективный обзор темы, предоставляя аргументы и контекст.
Статья предоставляет информацию, основанную на различных источниках и анализе.
Эта статья – источник ценной информации! Я оцениваю глубину исследования и разнообразие рассматриваемых аспектов. Она действительно расширила мои знания и помогла мне лучше понять тему. Большое спасибо автору за такую качественную работу!
Greetings from Florida! I’m bored to death at work so I decided to check out your website on my iphone during lunch break. I really like the information you provide here and can’t wait to take a look when I get home. I’m amazed at how quick your blog loaded on my cell phone .. I’m not even using WIFI, just 3G .. Anyhow, very good blog!
Статья помогла мне лучше понять сложные аспекты темы, с которыми я ранее не сталкивался.
Hey there superb website! Does running a blog like this require a lot of work? I have virtually no understanding of computer programming but I was hoping to start my own blog soon. Anyways, if you have any recommendations or techniques for new blog owners please share. I understand this is off subject however I just had to ask. Appreciate it!
Статья предлагает комплексный обзор событий, предоставляя различные точки зрения.
If some one wants to be updated with most recent technologies afterward he must be pay a quick visit this site and be up to date everyday.
Everyone loves what you guys tend to be up too. This sort of clever work and exposure! Keep up the superb works guys I’ve incorporated you guys to my personal blogroll.
Whats up very cool site!! Man .. Excellent .. Superb .. I will bookmark your website and take the feeds additionally? I’m satisfied to seek out so many useful info right here within the post, we want develop extra strategies in this regard, thanks for sharing. . . . . .
I’m gone to convey my little brother, that he should also pay a quick visit this blog on regular basis to take updated from most up-to-date news update.
Автор представляет сложные концепции и понятия в понятной и доступной форме.
Статья предлагает глубокий анализ проблемы, рассматривая ее со всех сторон.
Автор предлагает аргументы, подтвержденные достоверными источниками, чтобы убедить читателя в своих утверждениях.
I am not certain where you’re getting your information, but good topic. I must spend some time finding out much more or figuring out more. Thanks for magnificent info I used to be on the lookout for this information for my mission.
This information is worth everyone’s attention. Where can I find out more?
Статья содержит обширную информацию и аргументы, подтвержденные ссылками на достоверные источники.
Автор старается сохранить нейтральность, чтобы читатели могли основываться на объективной информации при формировании своего мнения. Это сообщение отправлено с сайта https://ru.gototop.ee/
Автор представляет сложные темы в понятной и доступной форме.
Hi to every one, for the reason that I am truly eager of reading this web site’s post to be updated regularly. It contains fastidious stuff.
Автор представляет аргументы с обоснованием и объективностью.
I absolutely love your website.. Very nice colors & theme. Did you make this website yourself? Please reply back as I’m attempting to create my very own site and would like to know where you got this from or exactly what the theme is named. Many thanks!
Автор старается быть объективным и предоставляет достаточно информации для осмысления и дальнейшего обсуждения.
Автор предоставляет актуальную информацию, которая помогает читателю быть в курсе последних событий и тенденций.
Я впечатлен этой статьей! Она не только информативна, но и вдохновляющая. Мне понравился подход автора к обсуждению темы, и я узнал много нового. Огромное спасибо за такую интересную и полезную статью!
Автор статьи представляет анализ и факты в балансированном ключе.
Автор старается сохранить нейтральность и предоставить балансированную информацию.
Информационная статья предлагает взвешенный подход к обсуждаемой теме, аргументируя свои выводы доказательствами и статистикой.
Hey there this is kind of of off topic but I was wanting to know if blogs use WYSIWYG editors or if you have to manually code with HTML. I’m starting a blog soon but have no coding expertise so I wanted to get guidance from someone with experience. Any help would be greatly appreciated!
Статья предлагает читателям широкий спектр информации, основанной на разных источниках.
Мне понравилась объективность и сбалансированность в подаче материала в статье.
Hi there just wanted to give you a quick heads up. The words in your content seem to be running off the screen in Chrome. I’m not sure if this is a formatting issue or something to do with internet browser compatibility but I thought I’d post to let you know. The layout look great though! Hope you get the issue resolved soon. Thanks
Я оцениваю широкий охват темы в статье.
I just like the valuable information you supply to your articles. I’ll bookmark your blog and take a look at again here regularly. I’m rather sure I will be informed many new stuff proper right here! Best of luck for the following!
Читателям предоставляется возможность самостоятельно рассмотреть и проанализировать информацию.
Читатели имеют возможность ознакомиться с разными точками зрения и самостоятельно оценить информацию.
Читателям предоставляется возможность ознакомиться с различными аспектами и сделать собственные выводы.
It’s amazing designed for me to have a web page, which is valuable in support of my experience. thanks admin
Автор старается оставаться объективным, чтобы читатели могли оценить различные аспекты и сформировать собственное понимание. Это сообщение отправлено с сайта https://ru.gototop.ee/
Статья содержит информацию, которая помогла мне расширить свои знания по данной теме.
Отличная статья! Я бы хотел отметить ясность и логичность, с которыми автор представил информацию. Это помогло мне легко понять сложные концепции. Большое спасибо за столь прекрасную работу!
I don’t even know how I ended up here, but I thought this post was good. I do not know who you are but certainly you are going to a famous blogger if you are not already 😉 Cheers!
Мне понравилось, как автор представил информацию в этой статье. Я чувствую, что стал более осведомленным о данной теме благодаря четкому изложению и интересным примерам. Безусловно рекомендую ее для прочтения!
Я оцениваю объективность и сбалансированность аргументации в статье.
Я нашел эту статью чрезвычайно познавательной и вдохновляющей. Автор обладает уникальной способностью объединять различные идеи и концепции, что делает его работу по-настоящему ценной и полезной.
Статья содержит достаточно информации для того, чтобы читатель мог получить общее представление о теме.
Эта статья просто великолепна! Она представляет информацию в полном объеме и включает в себя практические примеры и рекомендации. Я нашел ее очень полезной и вдохновляющей. Большое спасибо автору за такую выдающуюся работу!
Я бы хотел отметить качество исследования, проведенного автором этой статьи. Он представил обширный объем информации, подкрепленный надежными источниками. Очевидно, что автор проявил большую ответственность в подготовке этой работы.
Thankfulness to my father who shared with me about this website, this website is truly remarkable.
Hi! This is my 1st comment here so I just wanted to give a quick shout out and tell you I really enjoy reading through your posts. Can you recommend any other blogs/websites/forums that deal with the same subjects? Thank you!
My spouse and I absolutely love your blog and find almost all of your post’s to be exactly I’m looking for. can you offer guest writers to write content for you personally? I wouldn’t mind publishing a post or elaborating on a lot of the subjects you write about here. Again, awesome website!
Я хотел бы поблагодарить автора этой статьи за его основательное исследование и глубокий анализ. Он представил информацию с обширной перспективой и помог мне увидеть рассматриваемую тему с новой стороны. Очень впечатляюще!
Это помогает стимулировать обсуждение и критическое мышление у читателей.
Я нашел эту статью чрезвычайно познавательной и вдохновляющей. Автор обладает уникальной способностью объединять различные идеи и концепции, что делает его работу по-настоящему ценной и полезной.
Я нашел эту статью чрезвычайно познавательной и вдохновляющей. Автор обладает уникальной способностью объединять различные идеи и концепции, что делает его работу по-настоящему ценной и полезной.
Полезная информация для тех, кто стремится получить всестороннее представление.
Статья помогла мне получить глубокое понимание проблемы, о которой я раньше не задумывался.
It’s wonderful that you are getting thoughts from this paragraph as well as from our argument made at this place.
Pretty! This has been an extremely wonderful post. Many thanks for providing this info.
After going over a few of the blog articles on your website, I seriously appreciate your way of writing a blog. I book-marked it to my bookmark website list and will be checking back in the near future. Take a look at my web site as well and let me know how you feel.
Автор умело структурирует информацию, что помогает сохранить интерес читателя.
Статья помогла мне получить более полное представление о проблеме, которая рассматривается.
Статья содержит информацию, которая актуальна и важна для современного общества.
A motivating discussion is definitely worth comment. I do think that you should publish more about this issue, it might not be a taboo matter but typically people don’t discuss such topics. To the next! Many thanks!!
I absolutely love your blog.. Pleasant colors & theme. Did you develop this website yourself? Please reply back as I’m planning to create my own blog and would love to know where you got this from or just what the theme is named. Cheers!
I am now not certain where you’re getting your info, however great topic. I must spend some time learning much more or working out more. Thank you for great info I was in search of this info for my mission.
Надеюсь, что эти комментарии добавят ещё больше позитива и поддержки к информационной статье!
Pretty! This has been an extremely wonderful article. Thank you for supplying these details.
Мне понравился подход автора к представлению информации, он ясен и легко воспринимаем.
Hmm it appears like your website ate my first comment (it was super long) so I guess I’ll just sum it up what I wrote and say, I’m thoroughly enjoying your blog. I as well am an aspiring blog blogger but I’m still new to everything. Do you have any recommendations for novice blog writers? I’d really appreciate it.
Статья содержит разнообразные факты и аргументы, представленные в объективной манере.
Fantastic beat ! I would like to apprentice whilst you amend your site, how could i subscribe for a weblog website? The account helped me a appropriate deal. I had been tiny bit familiar of this your broadcast provided vibrant clear concept
Я восхищен глубиной исследования, которое автор провел для этой статьи. Его тщательный подход к фактам и анализу доказывает, что он настоящий эксперт в своей области. Большое спасибо за такую качественную работу!
Я хотел бы выразить признательность автору этой статьи за его объективный подход к теме. Он представил разные точки зрения и аргументы, что позволило мне получить полное представление о рассматриваемой проблеме. Очень впечатляюще!
Эта статья просто великолепна! Она представляет информацию в полном объеме и включает в себя практические примеры и рекомендации. Я нашел ее очень полезной и вдохновляющей. Большое спасибо автору за такую выдающуюся работу!
I really like it whenever people come together and share thoughts. Great website, stick with it!
Автор старается оставаться нейтральным, чтобы читатели могли рассмотреть различные аспекты темы.
Hi colleagues, how is the whole thing, and what you wish for to say on the topic of this piece of writing, in my view its actually awesome in favor of me.
Статья предоставляет объективную информацию о теме, подкрепленную различными источниками.
I’d like to find out more? I’d love to find out more details.
Link exchange is nothing else except it is simply placing the other person’s webpage link on your page at suitable place and other person will also do same in support of you.
It’s the best time to make a few plans for the future and it’s time to be happy. I’ve read this put up and if I may I want to recommend you some fascinating issues or tips. Perhaps you can write next articles relating to this article. I desire to learn more issues about it!
Автор старается предоставить объективную картину, оставляя читателям возможность самостоятельно сформировать свое мнение.
Hi, i think that i saw you visited my web site thus i came to “return the favor”.I’m attempting to find things to improve my site!I suppose its ok to use some of your ideas!!
Читателям предоставляется возможность ознакомиться с фактами и самостоятельно сделать выводы.
Статья представляет разнообразные аргументы и позиции, основанные на существующих данных и экспертном мнении.
Thank you, I’ve just been looking for information approximately this topic for a while and yours is the greatest I have came upon so far. But, what concerning the conclusion? Are you certain about the supply?
I was recommended this web site by my cousin. I’m not sure whether this post is written by him as no one else know such detailed about my problem. You’re wonderful! Thanks!
I’ve read a few excellent stuff here. Certainly price bookmarking for revisiting. I wonder how so much effort you set to make one of these excellent informative website.
Thanks for sharing your thoughts about meta_keyword. Regards
Автор статьи предоставляет информацию с разных сторон, представляя факты и аргументы.
Hello there! I know this is kinda off topic however , I’d figured I’d ask. Would you be interested in trading links or maybe guest writing a blog post or vice-versa? My website goes over a lot of the same topics as yours and I believe we could greatly benefit from each other. If you might be interested feel free to shoot me an email. I look forward to hearing from you! Fantastic blog by the way!
Автор старается предоставить объективную картину, оставляя читателям возможность самостоятельно сформировать свое мнение.
Pretty! This has been an extremely wonderful article. Many thanks for providing this information.
Hey I know this is off topic but I was wondering if you knew of any widgets I could add to my blog that automatically tweet my newest twitter updates. I’ve been looking for a plug-in like this for quite some time and was hoping maybe you would have some experience with something like this. Please let me know if you run into anything. I truly enjoy reading your blog and I look forward to your new updates.
Автор предоставляет примеры и иллюстрации, чтобы проиллюстрировать свои аргументы и упростить понимание темы.
Я хотел бы выразить свою благодарность автору этой статьи за исчерпывающую информацию, которую он предоставил. Я нашел ответы на многие свои вопросы и получил новые знания. Это действительно ценный ресурс!
Спасибо за эту статью! Она превзошла мои ожидания. Информация была представлена кратко и ясно, и я оставил эту статью с более глубоким пониманием темы. Отличная работа!
Мне понравилось, как автор представил информацию в этой статье. Я чувствую, что стал более осведомленным о данной теме благодаря четкому изложению и интересным примерам. Безусловно рекомендую ее для прочтения!
Hello it’s me, I am also visiting this website on a regular basis, this site is in fact good and the viewers are truly sharing pleasant thoughts.
Статья содержит актуальную статистику, что помогает более точно оценить ситуацию.
Статья предлагает читателю возможность самостоятельно сформировать свое мнение на основе представленных аргументов.
Автор не высказывает собственных предпочтений, что позволяет читателям самостоятельно сформировать свое мнение.
If some one wishes expert view about blogging then i suggest him/her to pay a quick visit this blog, Keep up the nice work.
Just want to say your article is as amazing. The clarity in your submit is just spectacular and that i could think you’re a professional on this subject. Fine with your permission let me to grab your RSS feed to keep updated with impending post. Thank you one million and please carry on the rewarding work.
Hi, Neat post. There is a problem along with your web site in internet explorer, could test this? IE still is the market leader and a good portion of other folks will leave out your magnificent writing due to this problem.
Статья содержит достаточно информации для того, чтобы читатель мог сделать собственные выводы.
Автор предоставляет примеры и иллюстрации, чтобы проиллюстрировать свои аргументы и упростить понимание темы.
Увеличение ссылочной массы: Ключевой фактор в росте DR сайта. Увеличение ссылочной массы является неотъемлемой частью стратегии роста DR сайта. Этот процесс требует систематического и длительного подхода, включающего в себя поиск высококачественных ссылок, мониторинг и анализ ссылочной массы, а также адаптацию к постоянно меняющемуся окружению поисковых систем. Понимание этих аспектов и их правильная реализация помогут сайту достичь более высокого DR, что в свою очередь улучшит его ранжирование, привлечет больше органического трафика и повысит успех онлайн-присутствия бизнеса.
Я благодарен автору этой статьи за его способность представить сложные концепции в доступной форме. Он использовал ясный и простой язык, что помогло мне легко усвоить материал. Большое спасибо за такое понятное изложение!
My brother recommended I might like this blog. He was totally right. This put up truly made my day. You cann’t believe just how much time I had spent for this info! Thanks!
Автор явно старается сохранить нейтральность и представить множество точек зрения на данную тему.
Автор старается оставаться нейтральным, предоставляя информацию для дальнейшего изучения.
Автор предоставляет различные точки зрения и аргументы, что помогает читателю получить полную картину проблемы.
I love what you guys are usually up too. This kind of clever work and reporting! Keep up the very good works guys I’ve included you guys to our blogroll.
Hi there! Quick question that’s entirely off topic. Do you know how to make your site mobile friendly? My weblog looks weird when browsing from my iphone. I’m trying to find a theme or plugin that might be able to fix this issue. If you have any recommendations, please share. Cheers!
Я хотел бы поблагодарить автора этой статьи за его основательное исследование и глубокий анализ. Он представил информацию с обширной перспективой и помог мне увидеть рассматриваемую тему с новой стороны. Очень впечатляюще!
Автор предлагает анализ плюсов и минусов разных подходов к решению проблемы.
I’m really loving the theme/design of your weblog. Do you ever run into any browser compatibility problems? A small number of my blog visitors have complained about my blog not working correctly in Explorer but looks great in Opera. Do you have any suggestions to help fix this issue?
I was able to find good advice from your content.
Автор представляет информацию в легком и доступном формате, что делает ее приятной для чтения.
Somebody essentially help to make critically articles I might state. This is the first time I frequented your web page and thus far? I surprised with the research you made to create this actual post amazing. Wonderful activity!
hello!,I love your writing very much! proportion we communicate extra approximately your post on AOL? I need a specialist on this space to unravel my problem. Maybe that’s you! Taking a look ahead to see you.
Я благодарен автору этой статьи за его тщательное и глубокое исследование. Он представил информацию с большой детализацией и аргументацией, что делает эту статью надежным источником знаний. Очень впечатляющая работа!
Hi there, I wish for to subscribe for this blog to obtain most recent updates, thus where can i do it please assist.
I got this site from my pal who told me about this website and now this time I am visiting this site and reading very informative articles or reviews at this place.
Статья представляет все основные аспекты темы, без излишней детализации.
Я ценю балансировку автора в описании проблемы. Он предлагает читателю достаточно аргументов и контекста для формирования собственного мнения, не внушая определенную точку зрения.
Статья помогла мне лучше понять сложные взаимосвязи в данной теме.
I’ve been surfing on-line greater than three hours these days, but I by no means discovered any interesting article like yours. It is lovely price enough for me. In my view, if all webmasters and bloggers made just right content material as you probably did, the web shall be much more helpful than ever before.
It’s perfect time to make some plans for the future and it is time to be happy. I’ve read this post and if I could I desire to suggest you few interesting things or suggestions. Maybe you can write next articles referring to this article. I want to read even more things about it!
Эта статья является примером качественного исследования и профессионализма. Автор предоставил нам широкий обзор темы и представил информацию с точки зрения эксперта. Очень важный вклад в популяризацию знаний!
At this time I am going away to do my breakfast, after having my breakfast coming yet again to read other news.
Статья ясно описывает факты и события, связанные с обсуждаемой темой.
Очень хорошо организованная статья! Автор умело структурировал информацию, что помогло мне легко следовать за ней. Я ценю его усилия в создании такого четкого и информативного материала.
I don’t even understand how I finished up here, but I assumed this submit was great. I don’t know who you might be but certainly you are going to a famous blogger if you aren’t already. Cheers!
Автор старается оставаться объективным, чтобы читатели могли сформировать свое собственное мнение на основе предоставленной информации.
Статья содержит аргументы, подкрепленные реальными примерами и исследованиями.
We’re a group of volunteers and starting a new scheme in our community. Your website offered us with valuable information to work on. You’ve performed an impressive job and our whole group will likely be thankful to you.
I like what you guys tend to be up too. This type of clever work and coverage! Keep up the superb works guys I’ve incorporated you guys to my own blogroll.
Автор статьи предоставляет разностороннюю информацию, основанную на различных источниках.
Статья предлагает глубокий анализ темы и рассматривает ее со всех сторон.
Это помогает читателям самостоятельно разобраться в сложной теме и сформировать собственное мнение.
Читатели имеют возможность самостоятельно проанализировать представленные факты и сделать собственные выводы.
Автор предлагает практические рекомендации, которые могут быть полезны в реальной жизни для решения проблемы.
Good day! Do you know if they make any plugins to safeguard against hackers? I’m kinda paranoid about losing everything I’ve worked hard on. Any tips?
Это помогает создать обстановку, в которой читатели могут обдумать и обсудить тему без пристрастия.
I was wondering if you ever thought of changing the page layout of your website? Its very well written; I love what youve got to say. But maybe you could a little more in the way of content so people could connect with it better. Youve got an awful lot of text for only having 1 or two pictures. Maybe you could space it out better?
Wow that was strange. I just wrote an very long comment but after I clicked submit my comment didn’t show up. Grrrr… well I’m not writing all that over again. Anyways, just wanted to say excellent blog!
Читателям предоставляется возможность оценить представленные данные и сделать собственные выводы.
Every weekend i used to go to see this web site, because i want enjoyment, since this this site conations in fact nice funny data too.
Attractive section of content. I just stumbled upon your website and in accession capital to assert that I acquire in fact enjoyed account your blog posts. Any way I’ll be subscribing to your augment and even I achievement you access consistently quickly.
My brother suggested I might like this blog. He was totally right. This post actually made my day. You cann’t imagine just how much time I had spent for this info! Thanks!
Автор предоставляет дополнительные ресурсы для тех, кто хочет углубиться в изучение темы.
Статья предоставляет информацию из разных источников, обеспечивая балансированное представление фактов и аргументов.
Pretty nice post. I just stumbled upon your blog and wished to say that I’ve truly enjoyed browsing your blog posts. After all I will be subscribing to your feed and I hope you write again very soon!
Автор предлагает обоснованные и логические выводы на основе представленных фактов и данных.
Я хотел бы отметить глубину исследования, представленную в этой статье. Автор не только предоставил факты, но и провел анализ их влияния и последствий. Это действительно ценный и информативный материал!
Надеюсь, вам понравятся эти комментарии! Это сообщение отправлено с сайта GoToTop.ee
Автор старается быть объективным и предоставляет достаточно информации для осмысления и дальнейшего обсуждения.
Статья содержит аргументы, подкрепленные реальными примерами и исследованиями.
I am regular reader, how are you everybody? This post posted at this site is genuinely pleasant.
Magnificent beat ! I wish to apprentice even as you amend your web site, how can i subscribe for a weblog web site? The account aided me a acceptable deal. I have been a little bit familiar of this your broadcast provided vivid clear concept
Hi! This is kind of off topic but I need some guidance from an established blog. Is it tough to set up your own blog? I’m not very techincal but I can figure things out pretty quick. I’m thinking about setting up my own but I’m not sure where to begin. Do you have any tips or suggestions? Thanks
Я оцениваю широту покрытия темы в статье.
I’m gone to convey my little brother, that he should also visit this weblog on regular basis to get updated from most recent information.
Мне понравилась объективность и сбалансированность в подаче материала в статье.
Я благодарен автору этой статьи за его тщательное и глубокое исследование. Он представил информацию с большой детализацией и аргументацией, что делает эту статью надежным источником знаний. Очень впечатляющая работа!
Полезная информация для тех, кто стремится получить всестороннее представление.
Это позволяет читателям самостоятельно сделать выводы и продолжить исследование по данному вопросу.
Автор старается подойти к теме нейтрально, предоставляя информацию, не влияющую на мнение читателей.
Автор предлагает анализ преимуществ и недостатков разных подходов к решению проблемы.
you’re in reality a excellent webmaster. The website loading speed is incredible. It kind of feels that you’re doing any distinctive trick. Moreover, The contents are masterwork. you have performed a magnificent process on this matter!
Hi, its good post concerning media print, we all understand media is a fantastic source of data.
Я хотел бы выразить свою благодарность автору этой статьи за исчерпывающую информацию, которую он предоставил. Я нашел ответы на многие свои вопросы и получил новые знания. Это действительно ценный ресурс!
My developer is trying to persuade me to move to .net from PHP. I have always disliked the idea because of the expenses. But he’s tryiong none the less. I’ve been using Movable-type on several websites for about a year and am concerned about switching to another platform. I have heard great things about blogengine.net. Is there a way I can transfer all my wordpress posts into it? Any kind of help would be really appreciated!
Статья содержит практические рекомендации, которые можно применить в реальной жизни для решения проблемы.
Have you ever thought about publishing an e-book or guest authoring on other sites? I have a blog based upon on the same topics you discuss and would love to have you share some stories/information. I know my visitors would appreciate your work. If you’re even remotely interested, feel free to send me an e-mail.
Автор предлагает логические выводы на основе представленных фактов и аргументов.
Я оцениваю широту покрытия темы в статье.
Я восхищен этой статьей! Она не только предоставляет информацию, но и вызывает у меня эмоциональный отклик. Автор умело передал свою страсть и вдохновение, что делает эту статью поистине превосходной.
Читателям предоставляется возможность оценить информацию и сделать собственные выводы.
Статья содержит ясные и убедительные аргументы, подкрепленные конкретными примерами.
Good post. I learn something totally new and challenging on websites I stumbleupon on a daily basis. It will always be interesting to read content from other authors and practice something from other websites.
It’s actually a cool and helpful piece of info. I am satisfied that you just shared this useful information with us. Please stay us informed like this. Thank you for sharing.
Мне понравилась организация информации в статье, которая делает ее легко восприимчивой.
Статья позволяет получить общую картину по данной теме.
Wow! Thank you! I continuously wanted to write on my website something like that. Can I take a part of your post to my blog?
Hey! I know this is kind of off topic but I was wondering which blog platform are you using for this website? I’m getting sick and tired of WordPress because I’ve had issues with hackers and I’m looking at options for another platform. I would be fantastic if you could point me in the direction of a good platform.
You need to take part in a contest for one of the most useful sites on the net.
I will highly recommend this site!
I pay a visit each day some websites and sites to read
articles or reviews, except this blog offers quality based content.
It’s an remarkable article designed for all the web
people; they will take advantage from it I am sure.
Hello.This post was extremely remarkable, especially because I was investigating for thoughts on this issue last Sunday.
You have mentioned very interesting points! ps nice web site. “He that will not sail till all dangers are over must never put to sea.” by Thomas Fuller.
This really answered my drawback, thank you!
Great post. I am facing a couple of these problems.
Thanks for sharing your info. I really appreciate your efforts
and I am waiting for your further post thanks once again.
Valuable information. Fortunate me I found your website by accident, and I am surprised
why this coincidence didn’t happened in advance!
I bookmarked it.
Hey there just wanted to give you a brief heads up and let you know a few of the pictures aren’t loading properly. I’m not sure why but I think its a linking issue. I’ve tried it in two different internet browsers and both show the same results.
An impressive share, I just given this onto a colleague who was doing a little analysis on this. And he in fact bought me breakfast because I found it for him.. smile. So let me reword that: Thnx for the treat! But yeah Thnkx for spending the time to discuss this, I feel strongly about it and love reading more on this topic. If possible, as you become expertise, would you mind updating your blog with more details? It is highly helpful for me. Big thumb up for this blog post!
I conceive other website proprietors should take this website as an example , very clean and superb user pleasant design and style.
Some genuinely nice and utilitarian info on this site, also I conceive the design has excellent features.
Es gibt viele solcher Diskussionen, die zeigen, dass es hilfreich ist, sich an den Kundenservice zu wenden.
Der Sicherheitsindex des Lord Lucky Casinos ist
überdurchschnittlich, und daher wird ein gründlicher Verifizierungsprozess durchgeführt, um sichere Transaktionen zu gewährleisten. Diese Hilfe ist besonders
nützlich, wenn technische Schwierigkeiten auftreten oder wenn Account-Verifizierungsprobleme während des KYC-Prozesses aufkommen. Falls die Probleme weiterhin bestehen, kann der Lord Lucky Kundenservice Ihnen weiterhelfen. Verwenden Sie in solchen Fällen die „Passwort vergessen“ Funktion auf der Login-Seite, um Ihre Anmeldedaten zurückzusetzen. Oftmals kann ein kleiner Tippfehler im Benutzernamen oder Passwort den Zugang verhindern.
Alle Funktionen wie Einzahlungen, Auszahlungen und der Kundensupport sind auch mobil verfügbar.
Ihre Daten sind durch 256-Bit-SSL-Verschlüsselung geschützt –
die gleiche Technologie, die Banken verwenden. Alle Spiele sind fair und werden regelmäßig von unabhängigen Prüforganisationen getestet.
Alle Auszahlungen werden innerhalb von 24 Stunden von unserem Team
genehmigt.
References:
https://online-spielhallen.de/die-sugar-casino-mobile-app-mein-umfassender-uberblick/
Eine vollständige Liste finden Sie in unseren Bonusbedingungen. Tischspiele wie Blackjack
und Roulette tragen zu 10% bei, Live-Casino-Spiele
zu 20%. Slots tragen zu 100% zur Erfüllung der Umsatzanforderungen bei.
Sie können das Bonusguthaben für fast alle unsere über 2.000 Spiele
verwenden. Kein Bonuscode erforderlich – alles läuft automatisch ab.
Ihre Bankdaten werden niemals auf unseren Servern gespeichert.
Vielversprechender als im Lord Lucky Casino kann der Name eines Online Casinos nicht sein. Bei Legale-Online-Casinos.de
bin ich unser Experte für Zahlungsanbieter Im Laufe der Zeit habe
ich dabei viele seriöse Casinos kennenlernen dürfen – aber eben auch einige schwarze
Schafe. Seit mehr als acht Jahren verfolge ich die Entwicklung insbesondere am deutschen Online-Glücksspielmarkt.
In der Casino-Branche bin ich mittlerweile ein alter Hase.
References:
https://online-spielhallen.de/verde-casino-aktionscode-dein-weg-zu-mehr-spielspas/
Coupled with the numerous game choices, this makes the welcome experience truly
memorable. This type of promotion allows users to test
out games without committing their own funds upfront. An appealing
treat for many newcomers is the Fair Go Casino no deposit bonus.
For those seeking deeper insights, a second Fair Go Casino review might reveal how offers evolve over time, catering to both new and returning gamblers.
The platform features high-payout percentages across all games, substantial jackpot pools exceeding
$1 million, and regular slot tournaments that create opportunities for significant wins.
For concerns related to the security of your profile,
only use support contacts found on the official Fair Go Casino website.
Fair Go Casino provides several responsive channels for resolving entry-related difficulties or recovery concerns, including those
affecting deposits or withdrawals in $.
Real Time Gaming provides the software that supports the
site, so you can expect to explore lots of their games if
you decide to become a member. How about exciting games to play, from table games to slots
and everything in between? This means you can receive a 100% match bonus
up to $200 on each of your first five deposits. Fair Go Casino welcomes new
Australian players with a generous welcome bonus package.
Join the chorus of satisfied winners today and discover why
Fair Go casino continues to dominate the Australian online gaming landscape.
The platform’s commitment to mobile optimization, responsive support, and player-first features demonstrates genuine dedication to customer satisfaction.
References:
https://blackcoin.co/casino-game-bank-craps/
Join thousands of Aussie players and grab your welcome bonus today.
Good news – the Rocket Casino mobile app offers a smooth, fast, and secure experience that’s tailor-made for Aussie users.
Massive selection of slots, table games, and live dealer
options from top providers.
No gimmicks, no grey areas, just pure gaming goodness. We’re not just
another flashy offshore casino. Whether you prefer live chat, email, or even smoke signals (okay, maybe not
that last one), help is always at hand.
It is important to emphasize that this commission does not affect the
bonuses or terms offered to you as a player. At Gambtopia.com, you’ll find a comprehensive overview of
everything worth knowing about online casinos. The bonuses are solid in size—with generous
deposit matches and frequent reloads—but the
45x wagering requirement is steeper than average. Yes, Casino Rocket fully supports AUD as both a deposit and playing currency.
References:
https://blackcoin.co/paypal-casinos-top-online-casinos-for-paypal-deposits-and-withdrawals/
FairGo Casino boasts a gaming experience to make your dreams come true in US-friendly online casino sites gaming.
From the no deposit bonus codes and welcome bonus coupons and codes to the fantastic collection of games,
this is one of the best casinos I was able to review.
Fair Go Casino does a fantastic job at creating a mobile platform
that enables players to bring the casino with them wherever they go.
RTG games are a huge hit amongst Aussie players
and FairGo knows how to get them sitting on the edge with RTG’s selection neatly offered to players.
Depending on the event, the tournament may focus on a specific
slot or a pool of games. Visa, Mastercard, Neosurf, CashtoCode, eZeeWallet, Skrill,
and Neteller are some payment options available
for deposits and withdrawals at Fair Go Casino.
The list of available payment methods at Fair Go Casino is
designed with every type of Aussie player in mind. It’s a simple reward for your loyalty,
giving you extra playtime without additional
deposits. Start every Thursday with a special
VIP bonus, delivered directly to your casino inbox.
First, Aussie players can enjoy a plethora of the most popular games in the land down under,
as well as generous Australian no deposit bonuses. While online
gambling laws vary by state, players can legally enjoy games at licensed international online casinos.
With its user-friendly design, secure payment options, and
24/7 customer support, it’s no wonder Fair Go has become a
favorite among Australian players.Ready to spin, win, and experience the thrill of online gaming?
As many online casinos employ their games, they provide the best progressive jackpots, which grow not just through our players but over
the internet.
References:
https://blackcoin.co/hellspin-australia-where-the-action-never-cools-down/
That’s why the casino offers a seamless mobile gaming experience
that allows you to enjoy your favorite games anywhere
across the vast Australian landscape – from busy city
centers to quiet coastal towns. Whether you’re playing pokies, table games,
or live dealer options, King Billy Casino delivers a seamless experience across desktop and mobile devices.
Online casino players can access a variety of games, including slots, table
games, and live dealer options.
Because in a crowded online gaming market, local touches—like accessible payment solutions and
tailored promotions—can make or break the experience.
That’s why online casino king billy australia typically partners with multiple software providers to keep the library fresh.
Whether you’re chasing progressive jackpots, exploring immersive live dealer tables, or spinning your
favorite pokies on mobile, King Billy delivers experiences that justify its growing reputation. Wagering requirements of 35x apply to bonus funds, while free spin winnings carry a 40x playthrough
before withdrawal. New players unlock a massive welcome package worth up to AU$2,500 plus 250 free spins
across four deposits.
References:
https://blackcoin.co/canasta-poker-rules-and-variations/
This post will assist the internet users for building up new weblog or even a blog
from start to end.
online betting with paypal winnersbet
References:
https://www.kondograpla.site/bbs/board.php?bo_table=free&wr_id=216
paypal casino online
References:
https://niazshomal.ir/city/babol/author/artlording0/
You made various fine points there. I did a search on the subject matter and found most people will consent with your blog.
online casino usa paypal
References:
https://jobs.ethio-academy.com/employer/the-best-new-online-casinos-in-australia-to-try-today/
online real casino paypal
References:
https://jobsonly.in/employer/best-welcome-bonuses-at-online-casinos-australia-in-2025/
Enjoyed reading through this, very good stuff, regards.
When I originally commented I clicked the “Notify me when new comments are added” checkbox and now each time a comment is added I get three emails with the same comment. Is there any way you can remove me from that service? Thanks!
Hello there, just became aware of your blog through Google, and found that it is
truly informative. I am gonna watch out for brussels.
I will appreciate if you continue this in future. Many people will be benefited from your writing.
Cheers!
Have you ever considered creating an ebook or guest authoring on other sites?
I have a blog centered on the same information you discuss and would
really like to have you share some stories/information. I know my visitors
would enjoy your work. If you are even remotely interested,
feel free to send me an e mail.
F*ckin’ amazing things here. I’m very happy to look your post. Thank you so much and i am having a look ahead to touch you. Will you kindly drop me a mail?
of course like your website but you have to test the spelling on several of
your posts. Several of them are rife with spelling problems and I to find it very bothersome to tell the reality then again I’ll certainly
come again again.
Definitely, what a splendid blog and informative posts, I surely will bookmark your site.All the Best!