承認を早期に取得するためには「申請戦略」から各設計文書を整備する必要がある
医療機器メーカーには、品目の承認または認証を早期に取得したいという要求があり、どうやれば申請がうまくいくか?早く通せるか?ということに注意が向くものだが、まずやらなければならないことは申請戦略を作ることである。
その申請戦略に基づいて、エビデンスを揃えて、申請資料を作らなければならないのである。
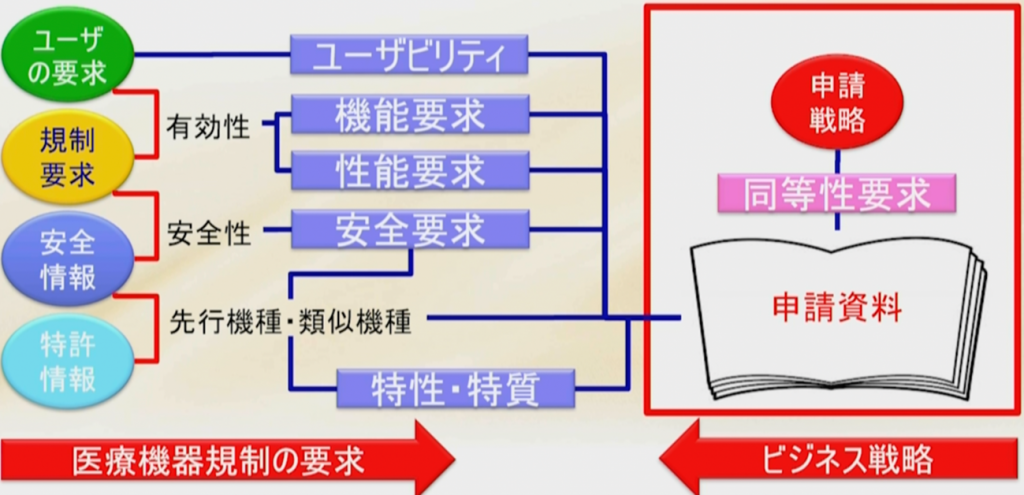
先の章で紹介したように、ユーザーの要求を集め、規制要求を集めて有効性を証明する、安全情報から来る安全要求で安全性を証明する。
さらに先行機種や類似機種に比べて特性・特質があるので、既に世の中に出ている機器または他社品、類似品に対してどんな特徴があるか特性があるかを出す。
新たな特性・特質により、前のものと性能の違う機能を持たせると新たにハザードが生まれ、新たに生まれるリスクをマネージメントする必要が出てくる。
少なくとも先行品に対して同等性を評価しなければならないし、同等以上のものでなければならない。
申請をする段になってこの試験が足りない、あの試験が足りないということにならないように、例えば改良医療機器として申請しようとしているのに新しい機能を付け加えてしまった、それによって新規医療機器になってしまったとか、クラスIIで抑えようと思ったのに、気がついたらクラスⅢになってしまっていた、などということがないようにしなければならない。
方針 (Step1) 〜申請資料〜
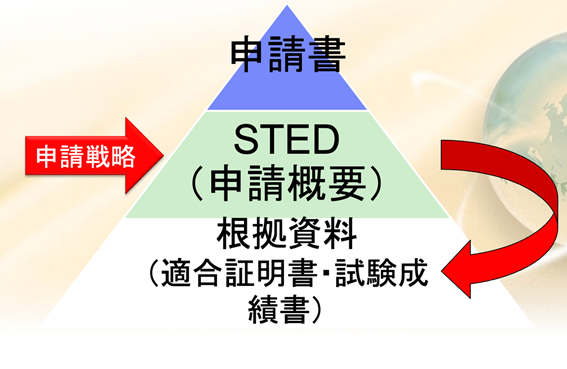
申請資料は、まず鏡として申請書がある。その下にステッドっていうものがある、これが申請概要である。
認証を受ける場合も承認を受ける場合も、このステッドを必ず書かなければならない。
このステッドというのは、サマリー文書のことをいう。
テクニカルドキュメントのサマリー文書は、日本語で言うと申請概要と言われるものである。
設計部門は、色々な設計資料、設計文書や仕様書であるとかまたはテストの記録、または外部試験機関による試験結果、成績証明書等を集めているが、それらすべてを認証申請の時に出すわけではない。
大事なことは、それらを集めた申請概要を書かなければならないということである
その申請概要の書き方、ステッドの書き方というのは当局から出ている。書き方の例というのがでているので、それを参照し作成する。
その根拠資料たるテクニカル文書(設計文書)は、いつ見られるかというと、申請をして、ステッドを見て。当局の方で信頼性調査を選ぶ。例えば二つ選ばれ、この資料を見せてください、次はこの資料を見せてくださいという風にランダムに選ばれる。どの資料を見せてくれと言われるか分からないので、すべての資料を取り揃えておかなければならない。
もし当局からの調査依頼に対して資料がなかった場合、慌てて作ったとしても間に合わない。医療機器の承認審査の期間というのは期限が決められており、その間に必要な資料が出せないと、申請が却下になってしまうということもあるので気をつけなければならない。
こういった根拠資料、多くの仕様書、そしてテスト結果、検証の記録、試験の成績書、これらの物を集めて、それをサマリーした文書がステッドと呼ばれるもので、このステッドを申請戦略に基づいて書くということが申請資料を作るということに相当する。
PMDA申請における調査対象
でここで気をつけていただきたいことなのですけど
ではどういう調査を受けるかということなのですね
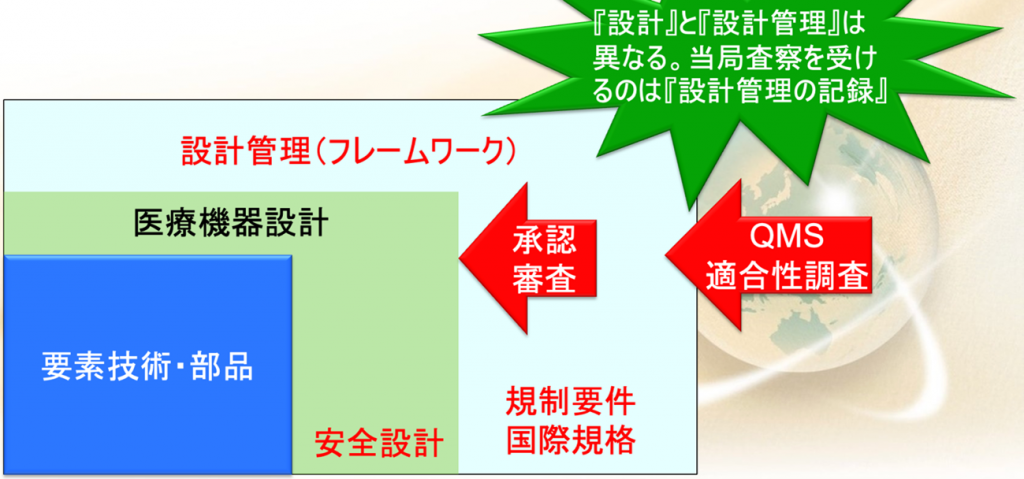
昨今は大学が要素技術を持っていることがある。
例えばソフトウェアのアルゴリズムだけあるなどするのだが、アルゴリズムだけあっても、実際には文書がないので、それでは設計したということにならない。
またもう一つは、この要素技術はどちらかといえば有効性の方で、安全設計はされてないことが多い。
そこで必要なのは図の青色に書いた要素技術をさらに拡大した、医療機器設計、医療機器に大事なことは安全設計なので、グリーンのエリアまで広めなければならないということなのだ。
さらに規制要件とか国際規格に沿ってその設計を管理しなければならない。
これを設計管理(フレームワーク)と言う
後述するが、設計と設計管理の違いを分かってない方が非常に多い。
設計と設計管理は異なるのである。
まず承認や認証で見られるのが、この医療機器設計の部分になる。
特に設計資料の中で、どんな機能で、どういう仕様で、どんな性能で、その結果がどうであって、ということと
もう一つは、安全設計が正しくされて、リスクが回避されている、リスクが低減されているかということである。
これを審査して、当該の医療機器は有効性・安全性がある、という結論になる。
で、もう一つ大事なことは、このQMS適合性調査というのが行われます
これは設計管理が対象になる。
設計と設計管理の違いについて述べる
医療機器の「設計」というのは、その医療機器そのものの有効性・安全性の審査のために必要となる。
一方で「設計管理」というのは、設計計画書を作ったか、計画書を遵守したか、結果が管理され、結果が記録として残っているか、当然内容が改ざんされていないこと、正しいこと
そういうことも管理しなければならない。
設計検証とかデザインレビューも設計審査が行われていて設計審査をする人が力量のない人では意味がない。素人がレビューしても決して品質の良い医療機器にはならないし、安全性の高い医療機器にならない。この設計審査に加わった人は誰であって、力量がどれくらいあるということを管理し記録に残っているかどうか、設計バリデーションを行って、臨床評価を行って、場合によっては臨床試験を行って、正しく管理されているかどうか、記録が残っているかどうか監査する。これをQMS適合性調査という。
「設計」は審査承認されるもの。「設計管理」はQMS適合性調査で査察されるもの、監査されるものという風にご理解いただきたい。
QMS省令で規制されているのは設計管理であって設計ではないので、そこをよく聞き分けていただきたい。
多くの場合は設計管理がうまくいってない。ISO-13485とかQSRとか、またはQMS省令に従った設計管理がうまくできていないということが多いので気をつけていただきたい。
日本の医療機器のクラス分類と承認要件・QMS調査要件
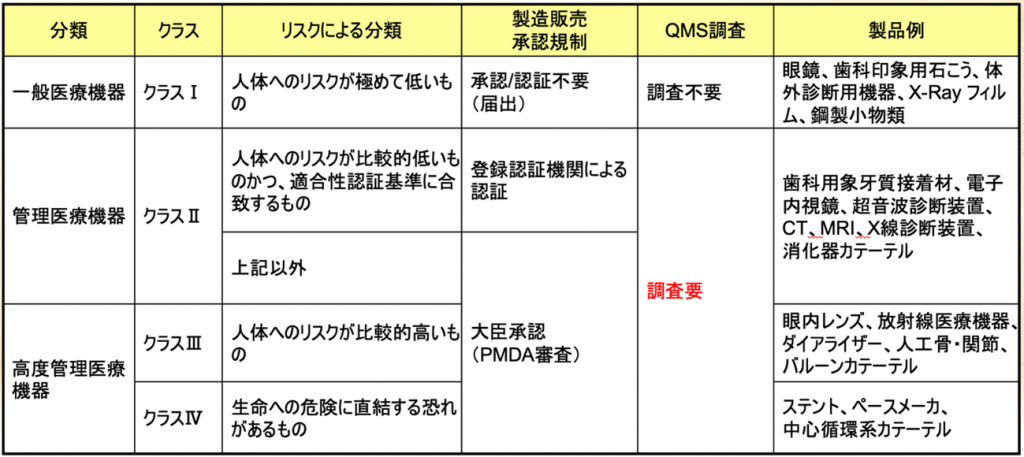
日本の医療機器のクラス分類と承認要件、QMS調査の要件を図でご覧いただきますと
一言です、クラスIを除けば、クラスIIとクラスⅢ、クラスⅣともにQMS適合性調査が必須であるということである。
2015年の薬事法の一部改正によって薬機法になったが、薬機法になって以降、QMS適合性調査は製販に入ることになった。
製販が製造業を監視監督していることを確認する。製販は各製造業者が持っている製品標準書集めて管理しておくことが原則である。
例えばメカは神奈川県で作っている、エレキは群馬県で作っている、ソフトウェアーは千葉県で作っているとする、そうするとメカだけ、エレキだけ、ソフトだけを監査をしても安全性・有効性が確認できない、品質が確認できない。医療機器として組上がって初めて品質が担保されているという事が確認できるので、QMSの適合性調査は製販に入るということになった。
製販に入って、そこから製造業者、これは主に最終組立工場(医療機器としてもう出荷すれば動くものに仕上げる工程のこと)、滅菌前でも相当するし、梱包とか滅菌前のもの、最終医療機器として動作する形にするところに対して実地調査が入ることがある。
実地調査があるかないかというのは当局のみぞ知るということなのだが、ISO-13485を取得していると実地調査が省略されることがある。もちろん省略されない場合も当然ある。
医療機器の民間の第三者機関による認証制度
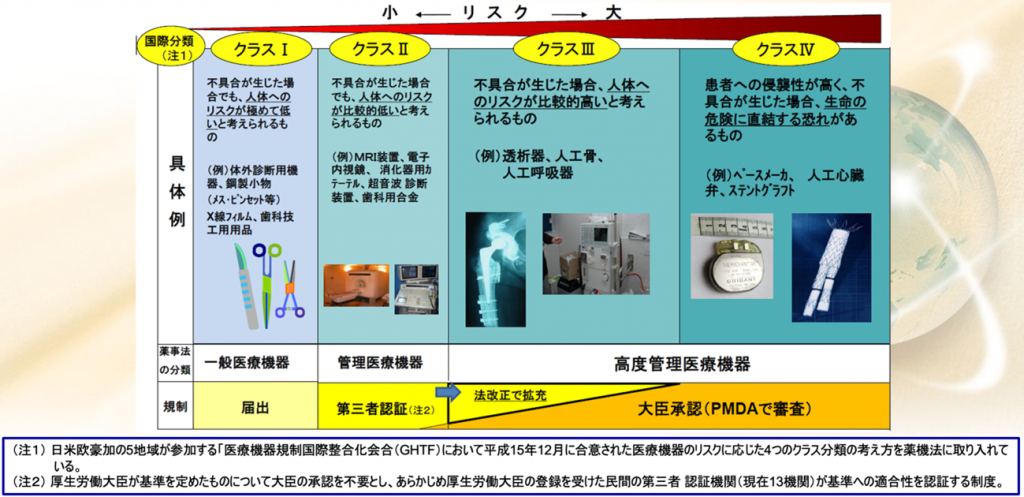
薬機法になってから、医療機器も承認権者が多少変わった。
品目に関しては、都道府県庁の監査というのはなくなった。
管理医療機器クラスIIは主に第三者認証になる。
クラスⅢは大臣承認ですから主にPMDAが審査することになっているが、一部のクラスIIは承認が必要になることがある。
逆に一部のクラスⅢの製品、例えば、もう枯れた製品、コンタクトレンズとかインプラントこういうものはクラスⅢ、要するに侵襲度が高かったり、接触時間が長かったりするのでクラスは高いけれども、安全性が十分に確認されているので第三者認証になったものもある。
一方で認証基準がない新規医療機器に関しては、クラスIIであってもPMDAの大臣承認
になるということもあり得る。日本はこのように承認権者が二つある。国・PMDAと第三者認証機関である。
アメリカは、ほぼ100%を当局FDAが審査をする。
一方でヨーロッパ100%第三者認証機関が認証する。しかもヨーロッパの場合は自己認証が基本で、当局は一切関与しない。
この違いは何かと言うと、承認権者が刑事訴追を受けるか受けないかなのである。
日本とアメリカは承認という制度があるのだが、その代わり、もしそれで万が一何らかの刑事訴追を患者の方から受けることになった場合は、国が成り代わって被告になるというのがこの承認という制度なのである。
一方ヨーロッパでは規制当局が一切この承認という行為に携わらない。 もし事故が起きた場合は当事者間で解決する。裁判を受けて立つというのがヨーロッパの制度なのだ。全て民間が認証するという制度になっており、日米とヨーロッパ欧州では考え方が全く違うということもご紹介しておきたい。
お役立ち動画
医療機器の設計・開発・申請における規制要件入門
~品質、有効性及び安全性の確保~
▶ 1講 医療機器と規制要件
▶ 2講 医療機器の種類
▶ 3講 医療機器と品質
▶ 4講 医療機器と安全性
▶ 5講 医療機器と有効性
▶ 6講 医療機器申請と当局査察
関連商品
[blogcard url=”https://ecompliance.co.jp/SHOP/QMS-LIVE.html” title=”(全16講)医療機器QMS規制入門セミナー【一括受講コース】” content=”医療機器企業に初めて入社・転職した人向けの規制要件入門コースです。QMSを中心に解説を行います。
本邦では、医療機器企業(製造販売業)においては、QMS省令(体制省令)およびGVP省令に則った体制の構築が出来ていなければなりません。
またQMS省令に従って、QMS(Quality Management System:品質管理システム)が文書化されており、適切にQMSに従って運用されており、それら活動の記録が作成されていなければなりません。
QMS省令は、ISO 13485をもとに作成されています。
ISO 13485は、マネジメント(経営者)の責任・リソース(資源)の配分(Plan)、製品実現(Do)、監視測定(Check)、改善(Action)といったPDCAサイクルで構成されています。
PDCAサイクルがあるということは、品質保証システムがあるということで、「今日」よりも「明日」、「明日」よりも「明後日」の方が品質が向上していくという証明となります。
規制当局は「継続的な改善」を求めています。
日進月歩の医療機器においては、常に設計変更が求められ、品質の改善も求められています。
本講座は16講(2時間/講)にわたって、医療機器業界にけるQMSに関する規制要件の内容を初心者にとって分かりやすく、丁寧に解説いたします。”]
[blogcard url=”https://ecompliance.co.jp/SHOP/QMS-MHLW-00.html” title=”【2021年度改正QMS省令対応】QMSひな形一式” content=”厚生労働省は、2021年3月26日付で「医療機器及び体外診断用医薬品の製造管理及び品質管理の基準に関する省令の一部を改正する省令」(令和3年厚生労働省令第60号)を公布しました。
経過措置期間は 3年間であり、2024年3月26日(改正省令の施行の日から起算して3年を経過する日)には新QMS省令を遵守しなければなりません。
今回の改正の趣旨は、QMS省令と医療機器の品質マネジメントシステムの国際規格であるISO13485:2016と整合を図ることです。
改正前のQMS省令はISO13485:2003年版と整合させており、最新の国際規格からは遅れていました。
厚生労働省は、2021年3月26日付で新QMS省令に関する逐条解説も公表しています。 「医療機器及び体外診断用医薬品の製造管理及び品質管理の基準に関する省令の一部改正について」(薬生発0326第10 号)
イーコンプライアンスでは、下記のお役立ち資料を作成し配布しております。
・改正QMS省令(本文)
・改正QMS省令と現行のQMS省令の対比表
・改正QMS省令逐条解説
・品質マニュアル(QMS省令2021年版対応)
・改正QMS省令手順化要求差異配布
・製品標準書新旧対応表配布
ご希望の方はこちらからダウンロードをお願いいたします。
すでに現行QMS省令に準拠したQMSを構築されている方で、改正対応が必要な方
これから医療機器に参入する方
にオススメのひな形セットです。”]
[blogcard url=https://xn--2lwu4a.jp/qms-md/ title=”QMS(手順書)ひな形 医療機器関連” ]
I love your blog.. very nice colors & theme. Did you create this website yourself? Plz reply back as I’m looking to create my own blog and would like to know wheere u got this from. thanks
I haven’t checked in here for some time because I thought it was getting boring, but the last several posts are good quality so I guess I will add you back to my daily bloglist. You deserve it my friend 🙂
I dugg some of you post as I thought they were very helpful very helpful
Great wordpress blog here.. It’s hard to find quality writing like yours these days. I really appreciate people like you! take care
Really superb info can be found on site.Raise blog range
Fantastic site. A lot of useful info here. I am sending it to several pals ans also sharing in delicious. And of course, thanks for your sweat!
Merely wanna say that this is invaluable, Thanks for taking your time to write this.
Some really interesting information, well written and broadly speaking user friendly.
Would you be serious about exchanging links?
The next time I read a blog, I hope that it doesnt disappoint me as much as this one. I mean, I know it was my choice to read, but I actually thought youd have something interesting to say. All I hear is a bunch of whining about something that you could fix if you werent too busy looking for attention.
Wow! Thank you! I continually wanted to write on my blog something like that. Can I implement a portion of your post to my blog?
I am glad to be a visitor of this staring blog! , thanks for this rare information! .
Sight Care
Fitspresso review
Cellucare review
Cellucare reviews
Lottery defeater
Sightcare reviews
Fitspresso
Sight Care
Nano defense pro
Fitspresso stands as a scientifically validated weight loss formula intricately crafted from potent ingredients, aimed at enhancing the body’s metabolic functions. Tailored to support the circadian rhythm and ignite the fat-burning process, this supplement is conveniently available in edible pill form for daily consumption.
Fitspresso
Cellu care
Cellu care
Sumatra Slim Belly Tonic reviews
Lottery defeater software
Fitspresso reviews
Fitspresso
Lottery defeater software reviews
Lottery defeater
Opini pix funciona
Opini pix é golpe
Ikaria Lean Belly Juice
Cellucare reviews
Fitspresso review
Fitspresso reviews
Fitspresso review
Fitspresso
Dentavim
Fitspresso
Fitspresso reviews
Fitspresso review
Fitspresso reviews
Fitspresso
Fitspresso
Fitspresso
Juice to loss weight
Rattling wonderful info can be found on web site. “The greatest mistake is trying to be more agreeable than you can be.” by Walter Bagehot.
Great remarkable issues here. I am very glad to peer your article. Thanks so much and i’m having a look forward to touch you. Will you please drop me a mail?
Zencortex reviews
Lottery defeater reviews
Neotonics gummies review
Sight Care reviews
Sight Care reviews
Dentavim
PROVADENT OFFICIAL
Dentavim is a dietary supplement formulated to support oral health and improve dental hygiene. With increasing awareness of the importance of maintaining good oral health, Dentavim has emerged as a popular choice for individuals seeking to enhance their dental care regimen. This article provides a detailed overview of Dentavim, including its ingredients, benefits, potential side effects, and overall effectiveness.
Lottery defeater software
But a smiling visitant here to share the love (:, btw outstanding design.
Nice blog! Is your theme custom made or did you download it from somewhere? A design like yours with a few simple adjustements would really make my blog shine. Please let me know where you got your design. Bless you
Sight Care reviews
Pineal XT reviews
Neotonics reviews
Kerassentials review
Hi, Neat post. There’s a problem with your website in web explorer, would check thisK IE still is the marketplace chief and a large element of people will omit your magnificent writing because of this problem.
sugar defender side effects
Cellucare reviews
Fitspresso review
sugar defender benefits
Fitspresso is a brand-new natural weight loss aid designed to work on the root cause of excess and unexplained weight gain. The supplement uses an advanced blend of vitamins, minerals, and antioxidants to support healthy weight loss by targeting the fat cells’ circadian rhythm
Javaburn reviews
Fitspresso review
Fitspresso reviews
Fitspresso
Fitspresso is a brand-new natural weight loss aid designed to work on the root cause of excess and unexplained weight gain. The supplement uses an advanced blend of vitamins, minerals, and antioxidants to support healthy weight loss by targeting the fat cells’ circadian rhythm
Fitspresso
Fitspresso is a brand-new natural weight loss aid designed to work on the root cause of excess and unexplained weight gain. The supplement uses an advanced blend of vitamins, minerals, and antioxidants to support healthy weight loss by targeting the fat cells’ circadian rhythm
Fitspresso
Java Burn
SUGAR DEFENDER REVIEWS
TONIC GREENS
sugar defender supplement
Lottery defeater software review
Lottery defeater software reviews
Fitspresso review
Fitspresso
Fitspresso review
Sight Care
Cellucare review
Puravive review
Lottery defeater review
Neotonics gummies reviews
Fitspresso reviews
Fitspresso review
LOTTERY DEFEATER SOFTWARE
LOTTERY DEFEATER
Nano defense pro reviews
Puravive
GROWTH MATRIX
Fitspresso review
Neotonics gummies reviews
Lift detox black
provadent review
SYNC SUPPLEMENT
PROVADENT REVIEW
LIPOZEM
LOTTERY DEFEATER REVIEWS
Lottery defeater system review
SUGAR DEFENDER REVIEWS
provadent supplement
The genius wave
LOTTERY DEFEATER SOFTWARE REVIEWS
SightCare
The genius wave system
Lottery defeater reviews
Lottery defeater system reviews
sugar defender supplement
Lottery defeater system review
The genius wave
Dentavim reviews
PROSTAVIVE REVIEWS
PLANTSULIN
Lipo slend reviews
Lipozem review
Puravive reviews
GLUCO FREEDOM REVIEWS
sugar defender
Provadent review
The genius wave
Dentavim review
Cellucare
Billionaire brain wave review
Erecprime review
LIPOZEM
ALPHA BITES REVIEWS
Pronerve
Lottery defeater reviews
ALPHA BITES
Dentavim reviews
Tonic Greens reviews
Pro nerve 6 reviews
Billionaire brain wave reviews
Sumatra Slim Belly Tonic review
Pineal XT review
Provadent review
Lottery defeater reviews
Plantsulin reviews
Liv care Liver support
Flexigenics reviews
Boostaro reviews
Lottery defeater software
Boostaro reviews
Fitspresso review
Sight Care
Lipozem review
The genius wave
ALPHA BITES REVIEW
Purevive reviews
Nanodefense pro reviews
PROSTAVIVE
Lipozem review
PROSTAVIVE REVIEWS
PROSTAVIVE
Boostaro reviews
Fitspresso
Alphabites review
Quietum plus reviews
nitric boost reviews
The genius wave review
Alpha bites
Lipozem reviews
PROVADENT REVIEW
SUGAR DEFENDER REVIEWS
Alphabites
LIPOZEM REVIEW
nitric boost results
Alphabites review
nagano reviews
Plantsulin review
Alphabites review
Lipozem reviews
Lottery defeater reviews
NAGANO TONIC
LOTTERY DEFEATER
ALPHA BITES
Alphabites review
Fitspresso review
Alpha bites
Alphabites reviews
LOTTERY DEFEATER SOFTWARE
BOOSTARO REVIEWS
FITSPRESSO REVIEW
LOTTERY DEFEATER
BOOSTARO
NAGANO TONIC
ALPHA BITES REVIEWS
Lottery defeater system reviews
Lipozem
Fitspresso
Alphabites
TURMERIC HACK
LIPOZEM REVIEW
PURAVIVE REVIEWS
FITSPRESSO
Hey there! Do you know if they make any plugins to protect against hackers? I’m kinda paranoid about losing everything I’ve worked hard on. Any recommendations?
Lipo Slim
Lipo Slim review
NERVOVIVE
You have brought up a very excellent details , regards for the post.
Alpha bites review
FREE SUGAR PRO REVIEW
Alphabites review
Lipozem review
Bariatric tea
Alpha bites review
Bariatric tea
Alpha bites
Alphabites
Femipro review
MANNA FLUX
Kerassentials
Alpha bites reviews
Boostaro review
TONIC GREENS
LEANBIOME REVIEW
Dentavim
SUGAR DEFENDER REVIEW
Booster XT ingredients
buy Booster XT
Alpha bites reviews
Alpha bites review
how does Booster XT work
Booster XT testosterone boost
NEUROQUIET
NEUROQUIET
Alpha bites reviews
Fitspresso review
Mitolyn
Lipozem reviews
Mitolyn review
Mitolyn
MITOLYN
Mitolyn reviews
Mitolyn
Mitolyn
buy Booster XT
Lottery defeater software
Fitspresso reviews
Alphabites
Alpha bites review
Nicoya PuraTea review
Booster XT testosterone boost
Mitolyn reviews
Mitolyn review
Mitolyn review
Mitolyn
Mitolyn
Mitolyn review
Mitolyn
Mitolyn
ALPHA BITES
MITOLYN REVIEWS
ALPHA BITES
Lipozem reviews
PROSTAZEN REVIEW
PROSTAZEN
LIPOZEM REVIEWS
SERENAFLOW REVIEWS
MITOLYN REVIEW
Mitolyn reviews
Mitolyn review
Fitspresso
Fitspresso review
Kerassentials review
MITOLYN REVIWS
TRIBALFORCE
MITOLYN
ALPHA BITES
Mitolyn
Mitolyn
Mitolyn
MITOLYN
Neuroquiet review
Mitolyn
Mitolyn
NEOTONICS REVIEW
SUGAR DEFENDER
Mitolyn review
Mitolyn
PROSTAZEN
PROSTAZEN REVIEWS
Nagano Lean Body Tonic review
Mitolyn
Leanbiome review
TRIBAL FORCE X REVIEW
TONIC GREENS REVIEWS
Mitolyn review
ProvaDent
TRIBAL FORCE X REVIEWS
MITOLYN
Mitolyn review
MITOLYN REVIEW
LeanBiome
LeanBiome review
Mitolyn review
LOTTERY DEFEATER SOFTWARE
LOTTERY DEFEATER SOFTWARE
Mitolyn review
Lottery Defeater review
ALPHA BITES REVIEWS
Mitolyn
PROVADENT
Mitolyn
Mitolyn review
Mitolyn reviews
Mitolyn
Mitolyn reviews
Mitolyn
Mitolyn reviews
Mitolyn
Mitolyn reviews
Mitolyn
MITOLYN REVIEW
MITOLYN
MITOLYN REVIEW
MITOLYN
MITOLYN REVIEWS
MITOLYN
Boostaro review
ERECPRIME REVIEW
Mitolyn
Tonic greens review
Tonic greens review
Quietum plus review
Nagano Lean Body Tonic
Nagano Lean Body Tonic
Nagano Lean Body Tonic Review
Mitolyn review
LOTTERY DEFEATER
ALPHA BITES
AIZEN POWER
LOTTERY DEFEATER REVIEWS
LOTTERY DEFEATER
AIZEN POWER REVIEWS
MITOLYN
MITOLYN
MITOLYN REVIEW
LOTTERY DEFEATER REVIEWS
DENTICORE REVIEWS
DENTICORE REVIEWS
PROSTAVIVE REVIEWS
PROSTAVIVE REVIEWS
TONIC GREENS REVIEWS
MITOLYN REVIEW
Mytolyn review
MITOLYN REVIEW
MITOLYN REVIEWS
MITOLYN
MITOLYN REVIEWS
MITOLYN REVIEWS
MITOLYN REVIEWS
MITOLYN REVIEWS
Mitolyn review
MITOLYN
MITOLYN REVIEW
MITOLYN
Alpha Bites
MITOLYN
MITOLYN REVIEW
MITOLYN REVIEWS
MITOLYN REVIEWS
MITOLYN REVIEW
MITOLYN REVIEWS
MITOLYN
SOULMATE STORY
Lottery Defeater
PRODENTIM
NATURAL MOUNJARO DRINK
Lottery Defeater
MITOLYN
NATURAL MOUNJARO DRINK
NATURAL MOUNJARO RECIPE
NATURAL MOUNJARO DRINK
Boostaro review
NATURAL MOUNJARO RECIPE
NATURAL MOUNJARO DRINK
NATURAL MOUNJARO DRINK
MITOLYN WEIGHT LOSS
Pronerve6 review
NATURAL MOUNJARO RECIPE
Billionaire Brain Wave Review
NATURAL MOUNJARO RECIPE
MITOLYN
MITOLYN
Billionaire Brain Wave Review
MITOLYN
MITOLYN REVIEW
MITOLYN REVIEWS
MITOLYN
MITOLYN
MITOLYN WEIGHT LOSS
MITOLYN REVIEW
MITOLYN REVIEW
Mitolyn
MITOLYN
MITOLYN REVIEW
Kerassentials
MITOLYN REVIEW
PRODENTIM REVIEWS
MITOLYN REVIEWS
MITOLYN REVIEWS
Boostaro review
MITOLYN REVIEWS
MITOLYN REVIEWS
Lottery Defeater Software
MITOLYN
NATURAL MOUNJARO RECIPE
MITOLYN REVIEWS
SUGAR DEFENDER
MITOLYN REVIEWS
Leanbiome
NATURAL MOUNJARO RECIPE
NAGANO TONIC REVIEWS
Mitolyn
MITOLYN REVIEWS
PRODENTIM REVIEWS
NAGANO TONIC
PRODENTIM REVIEWS
PRODENTIM REVIEW
PRODENTIM REVIEWS
NAGANO TONIC
NAGANO LEAN BODY TONIC
MITOLYN REVIEW
MITOLYN REVIEWS
NAGANO LEAN BODY TONIC
PRODENTIM REVIEW
NATURAL MOUNJARO
NATURAL MOUNJARO RECIPE 4 INGREDIENTS
NATURAL MOUNJARO RECIPE 4 INGREDIENTS
NATURAL MOUNJARO RECIPE 4 INGREDIENTS
NATURAL MOUNJARO RECIPE
POTENTSTREAM
NATURAL MOUNJARO
NATURAL MOUNJARO RECIPE 4 INGREDIENTS
NATURAL MOUNJARO
POTENT STREAM
NATURAL MOUNJARO RECIPE
NATURAL MOUNJARO
NATURAL MOUNJARO
NATURAL MOUNJARO RECIPE 4 INGREDIENTS
NATURAL MOUNJARO RECIPE 4 INGREDIENTS
NATURAL MOUNJARO RECIPE
POTENT STREAM
NATURAL MOUNJARO
NATURAL MOUNJARO
NATURAL MOUNJARO RECIPE 4 INGREDIENTS
POTENT STREAM REVIEWS
NATURAL MOUNJARO RECIPE
NATURAL MOUNJARO RECIPE
POTENT STREAM REVIEWS
NATURAL MOUNJARO RECIPE
MITOLYN REVIEW
MITOLYN REVIEWS
PROSTAZEN REVIEW
MITOLYN REVIEW
MITOLYN REVIEWS
PROSTAZEN REVIEWS
MITOLYN
MITOLYN REVIEWS
PROSTAZEN REVIEWS
MITOLYN REVIEW
MITOLYN REVIEWS
PROSTAZEN REVIEW
MITOLYN REVIEW
NATURAL MOUNJARO RECIPE
PROSTAZEN REVIEWS
NATURAL MOUNJARO
NATURAL MOUNJARO
PROSTAZEN
NATURAL MOUNJARO RECIPE
NATURAL MOUNJARO
PROSTAZEN
NATURAL MOUNJARO
NATURAL MOUNJARO RECIPE
NATURAL MOUNJARO RECIPE 4 INGREDIENTS
NATURAL MOUNJARO RECIPE
NATURAL MOUNJARO RECIPE 4 INGREDIENTS
Herpafend review
MITOLYN REVIEW
MITOLYN REVIEWS
Mitolyn
NITRIC BOOST
MITOLYN REVIEW
MITOLYN
NERVE FRESH REVIEW
MITOLYN
MITOLYN REVIEW
NERVE FRESH REVIEW
MITOLYN REVIEW
MITOLYN REVIEW
NERVE FRESH REVIEWS
MITOLYN
Java burn
MITOLYN REVIEW
NERVE FRESH REVIEWS
MITOLYN REVIEWS
Mitolyn
MITOLYN
NATURAL MOUNJARO
Mitolyn
MITOLYN REVIEWS
MITOLYN
Tea burn review
MITOLYN REVIEWS
KERASSENTIALS REVIEWS
MITOLYN
MITOLYN REVIEWS
MITOLYN
MITOLYN REVIEWS
MITOLYN
MITOLIN REVIEWS
MITOLYN REVIEW
MITOLYN
KERASSENTIALS
MITOLYN
SALT TRICK
SALT TRICK FOR MEN
SALT TRICK FOR MEN IN BED
SALT TRICK FOR MEN IN BED
SALT TRICK FOR MEN IN BED
SALT TRICK
SALT TRICK
SALT TRICK FOR MEN IN BED
SALT TRICK FOR MEN IN BED
SALT TRICK FOR MEN IN BED
SALT TRICK
SALT TRICK FOR MEN IN BED
SALT TRICK FOR MEN
SALT TRICK FOR MEN
SALT TRICK FOR MEN IN BED
SALT TRICK
SALT TRICK
SALT TRICK
SALT TRICK
Thank you ever so for you post.Much thanks again. Fantastic.
SALT TRICK FOR MEN
SALT TRICK FOR MEN
ALPHA BITES
ALPHA BITES
Mitolyn review
FITSPRESSO REVIEW
ALPHA BITES REVIEWS
ALPHA BITES REVIEW
ALPHA BITES REVIEW
Sight Care review
FITSPRESSO
ALPHA BITES REVIEWS
ALPHA BITES
Lottery Defeater
FITSPRESSO
NATURAL MOUNJARO
Leanbiome review
NATURAL MOUNJARO RECIPE 4 INGREDIENTS
SALT TRICK FOR MEN IN BED
MITOLYN REVIEWS
MITOLYN REVIEW
NATURAL MOUNJARO
Leanbiome review
Mitolyn
Mitolyn review
Mitolyn
Lottery Defeater
Lottery Defeater
Herpafend
Herpafend review
Prodentim review
wow, awesome article post. Really Cool.
Prodentim
Prodentim
SALT TRICK RECIPE
SALT TRICK RECIPE
SALT TRICK FOR MEN
LOTTERY DEFEATER
LOTTERY DEFEATER SOFTWARE
SALT TRICK
SALT TRICK RECIPE
SALT TRICK RECIPE
SALT TRICK
SALT TRICK
SALT TRICK
SALT TRICK RECIPE
SALT TRICK
SALT TRICK FOR MEN
NITRIC BOOST ULTRA
Boostaro review
NITRIC BOOST ULTRA REVIEWS
NITRIC BOOST
NITRIC BOOST ULTRA
NITRIC BOOST
Thanks for sharing, this is a fantastic post.Thanks Again.
Lottery defeater review
MITOLYN
MITOLYN
MITOLYN
Lottery defeater review
MITOLYN
Major thanks for the article. Fantastic.
NITRIC BOOST
NITRIC BOOST ULTRA
NITRIC BOOST ULTRA
Leanbiome
NITRIC BOOST ULTRA
NITRIC BOOST ULTRA
NITRIC BOOST ULTRA REVIEWS
NITRIC BOOST ULTRA
NITRIC BOOST
NITRIC BOOST
NITRIC BOOST ULTRA REVIEWS
My brother suggested I would possibly like this blog.He used to be entirely right. This put up truly made my day.You cann’t imagine simply how so much time I had spent for this information! Thanks!
NITRIC BOOST
MITOLYN
NATURAL MOUNJARO RECIPE
MITOLYN REVIEW
NATURAL MOUNJARO RECIPE
MITOLYN REVIEWS
MITOLYN
NATURAL MOUNJARO
MITOLYN REVIEW
NATURAL MOUNJARO
NATURAL MOUNJARO TEA
MITOLYN REVIEW
SALT TRICK FOR MEN
SALT TRICK FOR MEN
MITOLYN REVIEW
MITOLYN REVIEWS
MITOLYN REVIEWS
MITOLYN REVIEWS
MITOLYN REVIEWS
MITOLYN REVIEWS
MITOLYN REVIEWS
MITOLYN
MITOLYN
MITOLYN REVIEWS
Quietum Plus
MITOLYN
ZenCortex review
Quietum Plus review
Moundrops reviews
Tonic Greens
Moundrops reviews
Provadent review
Mitolyn reviews
TonicGreens
Mitolyn review
Tonic Greens
Mounjaro recipe
Tonic Greens review
Natural mounjaro recipe
Natural mounjaro
SALT TRICK
Salt trick for man growth
SALT TRICK RECIPE
SALT TRICK RECIPE
SALT TRICK RECIPE
Salt trick for men in bed
Salt trick to stay hard
Salt trick for man growth
Salt trick for man growth
SALT TRICK RECIPE
SALT TRICK FOR MEN
Salt trick explained
MITOLYN REVIEWS
MITOLYN
MITOLYN REVIEWS
MITOLYN
SALT TRICK
nitric boost ingredients
SALT TRICK FOR ED
SALT TRICK FOR ED
ultra boost
SALT TRICK FOR ED
SALT TRICK FOR MEN
nitric boost ingredients
SALT TRICK
SALT TRICK
SALT TRICK
SALT TRICK
SALT TRICK
SALT TRICK FOR MEN
SALT TRICK FOR ED
TURMERIC HACK
TURMERIC HACK FOR WEIGHT LOSS
TURMERIC HACK RECIPE
Mitolyn reviews
TURMERIC HACK
Mitolyn review
nitric boost ultra
SALT TRICK FOR MEN IN BED
Lottery Defeater
VERTIGENICS REVIEWS
SALT TRICK
Lean Body Tonic
SALT TRICK FOR MEN
Salt trick recipe
SALT TRICK
SALT TRICK FOR MEN IN BED
SALT TRICK FOR MEN
GlucoPure review
SALT TRICK FOR MEN
NITRIC BOOST ULTRA
NITRIC BOOST
Natural mounjaro
Looking forward to reading more. Great blog article.Much thanks again. Fantastic.
SALT TRICK FOR MEN
SALT TRICK FOR MEN
Neuroquiet review
SALT TRICK
MEMOFORCE REVIEWS
NITRIC BOOST
Salt trick for man growth
BLUE SALT TRICK
MEMOFORCE
SALT TRICK
NITRIC BOOST ULTRA
MEMOFORCE REVIEWS
SALT TRICK FOR MEN IN BED
SALT TRICK
Mitolyn review
MITOLYN REVIEWS
Neuroquiet
MITOLYN REVIEW
MITOLYN REVIEW
MITOLYN REVIEW
ICE WATER HACK
ICE HACK
ICE WATER HACK
Ozenvitta valor
ICE WATER DIET
ICE HACK
BLUE SALT TRICK
Java Burn
SALT TRICK FOR MEN
Salt Trick
BLUE SALT TRICK
SALT TRICK FOR MEN
nitric boost ultra review
SALT TRICK FOR MEN
NATURAL MOUNJARO RECIPE
SALT TRICK
nitric boost ultra
Salt Trick
SALT TRICK FOR MEN
BLUE SALT TRICK
Salt trick for men in bed
SALT TRICK FOR MEN
SALT TRICK FOR MEN
BLUE SALT TRICK
SALT TRICK FOR MEN
nitric boost ultra review
SALT TRICK
Mitolyn
SALT TRICK FOR MEN VIDEO
Salt trick for men in bed
SALT TRICK TO STAY HARD
nitric boost ultra
BLUE SALT TRICK FOR MEN IN BED
Leanbiome
SALT TRICK TUTORIAL
salt trick for men wrestling best
SALT TRICK TUTORIAL
BLUE SALT TRICK FOR MEN IN BED
SALT TRICK FOR MEN IN BED
ProDentim
SALT TRICK FOR MEN
SALT TRICK TUTORIAL
Boostaro
BRAZILIAN NATURAL MOUNJARO RECIPE
Boostaro review
BRAZILIAN MOUNJARO WEIGHT LOSS
BRAZILIAN MOUNJARO RECIPE
THE MONEY WAVE
BRAZILIAN MOUNJARO RECIPE
BRAZILIAN MOUNJARO DIET
BRAZILIAN MOUNJARO RECIPE
NATURAL MOUNJARO RECIPE
NATURAL MOUNJARO RECIPE
NATURAL MOUNJARO RECIPE
Lottery defeater
BRAZILIAN MOUNJARO DRINK
Nervefresh
BRAZILIAN MOUNJARO DRINK
SALT TRICK
Leanbiome
NATURAL MOUNJARO
ICE WATER HACK
Mitolyn
BLUE SALT TRICK
SALT TRICK FOR MEN
SALT TRICK FOR MEN
BLUE SALT TRICK
ICE WATER HACK DIET
SALT TRICK FOR MEN
SALT TRICK
ICE WATER HACK DIET
BLUE SALT TRICK
ICE WATER HACK
BLUE SALT TRICK
ICE WATER HACK TRICK
ICE WATER HACK
ICE WATER HACK
BLUE SALT TRICK
SALT TRICK
NATURAL MOUNJARO RECIPE
BLUE SALT TRICK
Zencortex
BLUE SALT TRICK
BLUE SALT TRICK
BLUE SALT TRICK
SALT TRICK
NATURAL MOUNJARO
BLUE SALT TRICK
BLUE SALT TRICK
Zencortex
BLUE SALT TRICK
BLUE SALT TRICK
SALT TRICK FOR MEN
BLUE SALT TRICK
Zencortex review
SALT TRICK FOR MEN
BLUE SALT TRICK
Sight care
SALT TRICK
SALT TRICK
Ice Water Hack for Weight Loss
SALT TRICK
SALT TRICK
SALT TRICK
SALT TRICK FOR MEN
ICE WATER HACK
BLUE SALT TRICK
Great post. I was checking continuously this blog and I’m impressed!Very helpful info specifically the last part 🙂 I care for such info much.I was looking for this particular information for a long time.Thank you and best of luck.
SALT TRICK
ICE WATER HACK
Sight care review
NANODEFENSE PRO
NANODEFENSE PRO
NATURAL MOUNJARO
Natural Mounjaro
BLUE SALT TRICK
SALT TRICK FOR MEN
SALT TRICK
NATURAL MOUNJARO
https://www.youtube.com/watch?v=A3RESE9jE90
Tonic Greens
SALT TRICK FOR MEN IN BED
SALT TRICK FOR MEN
SALT TRICK
SALT TRICK
SALT TRICK FOR MEN
SALT TRICK FOR MEN
SALT TRICK FOR MEN
SALT TRICK FOR MEN
SALT TRICK
SALT TRICK FOR MEN
SALT TRICK FOR MEN
SALT TRICK FOR MEN
MITOLYN REVIEW
MITOLYN
Sight care review
SALT TRICK
NATURAL MOUNJARO
MITOLYN REVIEWS
NATURAL MOUNJARO
Prodentim
SALT TRICK FOR MEN
NATURAL MOUNJARO
MITOLYN REVIEWS
Lottery Defeater
NATURAL MOUNJARO
ICE WATER HACK
ICE WATER HACK FOR WEIGHT LOSS
SALT TRICK
NATURAL MOUNJARO
SALT TRICK
SALT TRICK
SALT TRICK FOR MEN
TURMERIC TRICK
SALT TRICK
SALT TRICK
SALT TRICK FOR MEN
SALT TRICK
SALT TRICK
BLUE SALT TRICK
SALT TRICK
BLUE SALT TRICK
SALT TRICK
SALT TRICK
SALT TRICK
SALT TRICK FOR MEN
SALT TRICK FOR MEN
SALT TRICK
SALT TRICK
Mitolyn
NATURAL MOUNJARO
SALT TRICK FOR MEN
SALT TRICK FOR MEN
SALT TRICK
SALT TRICK
SALT TRICK
SALT TRICK
SALT TRICK
SALT TRICK FOR MEN
SALT TRICK FOR MEN
SALT TRICK
SALT TRICK
SALT TRICK FOR MEN
SALT TRICK FOR MEN
SALT TRICK FOR MEN
SALT TRICK
SALT TRICK FOR MEN
SALT TRICK FOR MEN
ICE WATER HACK WEIGHT LOSS
SALT TRICK
ICE WATER HACK
SALT TRICK FOR MEN
ICE WATER HACK WEIGHT LOSS
SALT TRICK FOR MEN
Tonic Greens
SALT TRICK
ICE HACK DIET
ICE WATER HACK
SALT TRICK
SALT TRICK
ICE WATER HACK
ICE HACK RECIPE
SALT TRICK
SALT TRICK
SALT TRICK
Sight care
LOTTO CHAMP REVIEWS
SALT TRICK FOR MEN
LOTTOCHAMP
SALT TRICK FOR MEN
SALT TRICK FOR MEN
LOTTOCHAMP
Sight care
Tonic Greens review
Mitolyn
Lottery defeater review
Lottery defeater
Boostaro
SALT TRICK FOR MEN
TURMERIC HACK RECIPE
SALT TRICK FOR MEN
TURMERIC HACK FOR WEIGHT LOSS
Natural Mounjaro recipe
Natural Mounjaro
TURMERIC HACK TO WEIGHT LOSS
TURMERIC HACK RECIPE
TURMERIC HACK
COFFEE LOOPHOLE RECIPE
Sight care review
Java Burn
SALT TRICK FOR MEN
Prodentim
SALT TRICK
Prodentim review
Leanbiome
Leanbiome
Prodentim
Zencortex
Boostaro
Sight care review
NATURAL MOUNJARO RECIPE
Sight care review
LOTTERY DEFEATER SOFTWARE
Nagano Lean Body Tonic
SALT TRICK FOR MEN
SALT TRICK FOR MEN
SALT TRICK
SALT TRICK
NATURAL MOUNJARO
SALT TRICK
SALT TRICK FOR MEN
Lottery Defeater
NeuroQuiet review
Thanks for the post.Thanks Again. Really Great.
Quietum Plus review
Sight care
Prodentim review
Prodentim review
MITOLYN REVIEWS
SALT TRICK
I cannot thank you enough for the article post.Really thank you! Great.
SALT TRICK FOR MEN
SALT TRICK FOR MEN
BRAZILIAN MOUNJARO RECIPE
NATURAL MOUNJARO RECIPE
Leanbiome review
Aqua Sculpt
LIPOZEM REVIEW
SALT TRICK
ICE WATER HACK
SALT TRICK FOR MEN
LIPOZEM REVIEW
LIPOZEM REVIEWS
Aqua Sculpt
Lottochamp
ICE WATER HACK RECIPE
ICE WATER HACK FOR WEIGHT LOSS
ICE WATER HACK
SALT TRICK FOR MEN
BLUE SALT TRICK
Major thanks for the blog.Thanks Again. Keep writing.
Prostazen review
BLUE SALT TRICK
SALT TRICK FOR MEN
SALT TRICK FOR MEN
WHISPEARA
SALT TRICK
Gluco Extend
Prime Biome
NATURAL MOUNJARO RECIPE
Gluco Tonic
Gluco Tonic
Sight care
I appreciate you sharing this blog article.Thanks Again. Really Cool.
Vertigenics
Appanail
Prodentim review
SALT TRICK
SALT TRICK
Zencortex
SALT TRICK FOR MEN
BRAZILIAN MOUNJARO RECIPE
BRAZILIAN MOUNJARO RECIPE
https://www.youtube.com/watch?v=7IWocKZbVq0
SALT TRICK
Denticore
Nerve fresh review
Moringa magic
PRODENTIM
THE GROWTH MATRIX
Salt trick for man growth
Salt trick for man
Boostaro review
Salt trick for men
Lottery defeater review
Salt trick for man growth
Awesome blog article.Really thank you! Fantastic.
ICE WATER HACK
Prodentim review
Ice water hack diet
Prodentim review
ICE WATER HACK FOR WEIGHT LOSS
MITOLYN REVIEW
MITOLYN REVIEWS
ICE WATER HACK FOR WEIGHT LOSS
Gluco6
Nitric Boost
Nitric boost ultra reviews
Nitric boost ultra reviews
ICE WATER HACK
Salt trick for men
Prodentim
BLACK WOOD TEA
Quietum plus review
primebiome review
primebiome reviews
primebiome review
I value the blog article. Want more.
primebiome
LeanBiome review
BRAZILIAN MOUNJARO
Im thankful for the article.Really looking forward to read more. Cool.
Prodentim review
Natural mounjaro
PRIME BIOME REVIEW
Brazilian natural mounjaro
PINK SALT TRICK RECIPE
Prodentim
Natural mounjaro recipe
Prodentim
Lottochamp
Natural mounjaro recipe
BRAZILIAN MOUJARO RECIPE
Natural mounjaro recipe
Brazilian natural mounjaro
Mitolyn
Lottochamp
NATURAL MOUNJARO
Natural mounjaro
Lotto Champ review
Mitolyn review
Natural mounjaro recipe
MITOLYN
Tonic Greens review
Natural Mounjaro
Natural Mounjaro
BLUE SALT TRICK
Very neat blog. Much obliged.
BRAZILIAN MOUNJARO RECIPE
Great blog.Really thank you! Want more.
The design and usability are top-notch, making everything flow smoothly.
PRODENTIM REVIEW
The design and usability are top-notch, making everything flow smoothly.
I’m really impressed by the speed and responsiveness.
I love how user-friendly and intuitive everything feels.
Very neat blog.Thanks Again. Really Great.
The content is engaging and well-structured, keeping visitors interested.
I value the blog post.Really looking forward to read more. Fantastic.
Major thankies for the blog article.Thanks Again. Really Great.
The content is well-organized and highly informative.
Very neat blog article.Much thanks again. Will read on…
The content is engaging and well-structured, keeping visitors interested.
A perfect blend of aesthetics and functionality makes browsing a pleasure.
I value the blog post.Much thanks again. Awesome.
I love how user-friendly and intuitive everything feels.
I’m really impressed by the speed and responsiveness.
The content is engaging and well-structured, keeping visitors interested.
The content is engaging and well-structured, keeping visitors interested.
This site truly stands out as a great example of quality web design and performance.
Muchos Gracias for your article post.Really looking forward to read more. Will read on…
The content is engaging and well-structured, keeping visitors interested.
The design and usability are top-notch, making everything flow smoothly.
The content is well-organized and highly informative.
Nothing beats homemade pasta. The texture and flavor are just on another level compared to store-bought versions. Cooking from scratch is truly an art.
Consistency is key in fitness. You won’t see results overnight, but every workout counts. The small efforts add up over time and create real change.
Live concerts have a special magic. No recording can ever capture that raw energy of the crowd and the artist performing in the moment.
Watching a sunset over the ocean is one of the most peaceful experiences in life. Nature has a way of reminding us how small but connected we all are.
I value the post.Really thank you! Cool.
Thanks-a-mundo for the article. Keep writing.
Really informative blog.Much thanks again. Really Cool.
Muchos Gracias for your post.Really looking forward to read more. Keep writing.
I appreciate you sharing this post.Really thank you! Great.
All knowledge, it is said, comes from experience, but does that not mean that the more we experience, the wiser we become? If wisdom is the understanding of life, then should we not chase every experience we can, taste every flavor, walk every path, and embrace every feeling? Perhaps the greatest tragedy is to live cautiously, never fully opening oneself to the richness of being.
Man is said to seek happiness above all else, but what if true happiness comes only when we stop searching for it? It is like trying to catch the wind with our hands—the harder we try, the more it slips through our fingers. Perhaps happiness is not a destination but a state of allowing, of surrendering to the present and realizing that we already have everything we need.
The cosmos is said to be an ordered place, ruled by laws and principles, yet within that order exists chaos, unpredictability, and the unexpected. Perhaps true balance is not about eliminating chaos but embracing it, learning to see the beauty in disorder, the harmony within the unpredictable. Maybe to truly understand the universe, we must stop trying to control it and simply become one with its rhythm.
The potential within all things is a mystery that fascinates me endlessly. A tiny seed already contains within it the entire blueprint of a towering tree, waiting for the right moment to emerge. Does the seed know what it will become? Do we? Or are we all simply waiting for the right conditions to awaken into what we have always been destined to be?
Virtue, they say, lies in the middle, but who among us can truly say where the middle is? Is it a fixed point, or does it shift with time, perception, and context? Perhaps the middle is not a place but a way of moving, a constant balancing act between excess and deficiency. Maybe to be virtuous is not to reach the middle but to dance around it with grace.
Virtue, they say, lies in the middle, but who among us can truly say where the middle is? Is it a fixed point, or does it shift with time, perception, and context? Perhaps the middle is not a place but a way of moving, a constant balancing act between excess and deficiency. Maybe to be virtuous is not to reach the middle but to dance around it with grace.
The cosmos is said to be an ordered place, ruled by laws and principles, yet within that order exists chaos, unpredictability, and the unexpected. Perhaps true balance is not about eliminating chaos but embracing it, learning to see the beauty in disorder, the harmony within the unpredictable. Maybe to truly understand the universe, we must stop trying to control it and simply become one with its rhythm.
The potential within all things is a mystery that fascinates me endlessly. A tiny seed already contains within it the entire blueprint of a towering tree, waiting for the right moment to emerge. Does the seed know what it will become? Do we? Or are we all simply waiting for the right conditions to awaken into what we have always been destined to be?
Thanks a lot for the blog. Really Great.
The essence of existence is like smoke, always shifting, always changing, yet somehow always present. It moves with the wind of thought, expanding and contracting, never quite settling but never truly disappearing. Perhaps to exist is simply to flow, to let oneself be carried by the great current of being without resistance.
If everything in this universe has a cause, then surely the cause of my hunger must be the divine order of things aligning to guide me toward the ultimate pleasure of a well-timed meal. Could it be that desire itself is a cosmic signal, a way for nature to communicate with us, pushing us toward the fulfillment of our potential? Perhaps the true philosopher is not the one who ignores his desires, but the one who understands their deeper meaning.
Thanks a lot for the blog post.Much thanks again. Really Cool.
Thanks a lot for the article post. Really Great.
Very good blog post.Thanks Again. Much obliged.
Lottochamp
Prodentim
Appanail review
SALT TRICK FOR MEN
Prime biome
Appanail review
SALT TRICK FOR MAN
Jangan berhenti berbagi ya, karena artikel seperti ini sangat dinantikan sama komunitas pecinta slot kayak saya.
WHISPEARA REVIEW
slot deposit pulsa tanpa potongan – Sekali lagi, makasih banyak buat penulis yang udah ngasih artikel sekeren ini. Jangan berhenti berbagi ya, karena artikel seperti ini sangat dinantikan sama komunitas pecinta slot kayak saya. Sukses terus dan semoga makin banyak pemain yang terbantu karena artikel ini!
slot deposit pulsa tanpa potongan – Sekali lagi, makasih banyak buat penulis yang udah ngasih artikel sekeren ini. Jangan berhenti berbagi ya, karena artikel seperti ini sangat dibutuhkan sama komunitas pecinta slot kayak saya. Sukses terus dan semoga makin banyak pemain yang terbantu karena artikel ini!
Herpafend Review
I really liked your blog article. Fantastic.
Prodentim
Prodentim
Leanbiome
Tonic greens review
slot gacor hari ini – Saya pribadi sangat tertarik oleh gaya penyampaian artikel ini. Penjelasannya to the point dan langsung menyasar ke inti pembahasan, bikin makin percaya buat daftar di situs slot gacor yang dibahas.
NerveFresh
NATURAL MOUNJARO RECIPE
NATURAL MOUNJARO RECIPE
The cosmos is said to be an ordered place, ruled by laws and principles, yet within that order exists chaos, unpredictability, and the unexpected. Perhaps true balance is not about eliminating chaos but embracing it, learning to see the beauty in disorder, the harmony within the unpredictable. Maybe to truly understand the universe, we must stop trying to control it and simply become one with its rhythm.
Gluco Tonic
Gluco Tonic review
ELEPHANT ROOT TRICK
Prodentim review
The cosmos is said to be an ordered place, ruled by laws and principles, yet within that order exists chaos, unpredictability, and the unexpected. Perhaps true balance is not about eliminating chaos but embracing it, learning to see the beauty in disorder, the harmony within the unpredictable. Maybe to truly understand the universe, we must stop trying to control it and simply become one with its rhythm.
Zencortez
Prime Biome
Salt Trick review
Quietum plus
Prodentim
I couldn’t resist commenting
Appanail
Say, you got a nice blog. Fantastic.
Slimberine Review
ARIALIEF
Salt trick review
Nitric Boost review
herpafend
NERVE FRESH REVIEW
Boostaro review
Lottery defeater review
Slimjaro
Mitolyn review
Gluco6 review
Major thanks for the blog.Really looking forward to read more. Really Cool.
Beast Force
Gluco control review
Im thankful for the post.Thanks Again. Great.
ELEPHANT ROOT TRICK
I really liked your blog.Really thank you! Will read on…
Neuroquiet
A round of applause for your blog post.Really thank you! Want more.
Provadent review
Say, you got a nice article post.Thanks Again. Keep writing.
slot gacor – Postingan ini benar-benar menginspirasi dan mampu menambah pengetahuan seputar dunia judi slot digital yang sedang trend. Terima kasih atas pengalaman yang komprehensif, cocok banget untuk penggemar slot seperti saya yang lagi cari slot cuan.
Gumaktiv review
slot gacor – Postingan ini benar-benar bermanfaat dan mampu membuka wawasan seputar dunia slot online yang sedang naik daun. Terima kasih atas pengalaman yang lengkap, cocok banget untuk pencari RTP tinggi seperti saya yang lagi cari slot cuan.
Enjoyed every bit of your article post.Really thank you! Awesome.
Leanbiome
Prostavive
Gluco Extend review
The essence of existence is like smoke, always shifting, always changing, yet somehow always present. It moves with the wind of thought, expanding and contracting, never quite settling but never truly disappearing. Perhaps to exist is simply to flow, to let oneself be carried by the great current of being without resistance.
Beastforce
Lean Body Tonic
Lottery Defeater review
Mitolyn
Glucotrust
Billionaire Brain Ware
Billionaire Brain Ware Review
Kerassentials review
Herpesyl
Fitspresso
Tonic greens review
Dentatonic
Aw, this was a very nice post. Taking the time and actual effort to produce a very good articleÖ but what can I sayÖ I procrastinate a whole lot and don’t manage to get anything done.
Manganese resembles iron in its chemical and physical residential properties, however it is harder and also extra brittle. Manganese is possibly one of the most flexible aspect that can be added to copper alloys.
Wow, great blog post.Really thank you! Really Great.
I am so grateful for your article post.Really thank you! Really Cool.
It?s tough to locate knowledgeable people on this topic, but you seem like you recognize what you?re speaking about! Thanks
We are looking for experienced people that might be interested in from working their home on a full-time basis. If you want to earn $500 a day, and you don’t mind developing some short opinions up, this is the perfect opportunity for you!
A big thank you for your post.Thanks Again. Will read on…
Thanks-a-mundo for the article.Thanks Again. Great.
Hey, thanks for the blog post.Thanks Again. Will read on…
I enjoy looking through an article that will make men and women think. Also, thanks for permitting me to comment!
Great, thanks for sharing this blog. Really Cool.
Is anyone here in a position to recommend Hollow Butt Plugs? Thanks xx
I think this is a real great blog post. Really Cool.
Appreciate you sharing, great blog.Thanks Again.
Thanks again for the blog.Thanks Again. Fantastic.
Thank you ever so for you post.Really looking forward to read more. Great.
Im grateful for the article.Thanks Again.
Very good blog article.Really thank you! Fantastic.
This very blog is no doubt cool and informative. I have chosen many interesting things out of this blog. I ad love to visit it again soon. Thanks a bunch!
Xoilac Tv Thẳng Bóng Đá xem da bong truc tiep k+Đội tuyển chọn futsal nước ta đã được một trận đấu đồng ý được trước đối thủ cạnh tranh đầy mức độ mạnh Lebanon. Kết quả Bà Rịa-Vũng Tàu vs Bình Phước hôm nay 18h00 ngày 5/5, Hạng nhất nước Việt Nam.
Lift detox caps
Wow! This can be one particular of the most beneficial blogs We have ever arrive across on this subject. Actually Excellent. I’m also an expert in this topic therefore I can understand your hard work.
Im thankful for the post.Really thank you! Fantastic.
Hi there, after reading this amazing piece of writing i am as well glad to sharemy experience here with friends.
An interesting discussion is definitely worth comment. I do think that you ought to write more on this topic, it might not be a taboo matter but typically people do not speak about such topics. To the next! All the best!!
Im thankful for the article post.Thanks Again.
ivermectin germany preço do stromectol Opmuhf42 ivermectin generic
Thanks for your marvelous posting! I genuinely enjoyed reading it, you are a great author.I will be sure to bookmark your blog and definitely will come back someday. I want to encourage one to continue your great job, have a nice weekend!
I’m not sure where you are getting your information, but good topic. I needs to spend some time learning more or understanding more. Thanks for great info I was looking for this information for my mission.
I just like the helpful information you provide for your articles. I’ll bookmark your blog and check once more right here regularly. I am reasonably sure I’ll be informed lots of new stuff proper right here! Best of luck for the next!
I really like and appreciate your article post.
I truly appreciate this blog post.Thanks Again. Will read on…
how to write an essayclassification essayhelp writing a essay
Thanks so much for the blog article.Really looking forward to read more.
I don’t even understand how I stopped up here, howeverI believed this post used to be good. I do not understand who you are however definitely you’re going to a well-known bloggerin case you are not already. Cheers!
A big thank you for your blog.Much thanks again. Awesome.
What’s up i am kavin, its my first occasion to commenting anywhere, when i read this post i thought i could also createcomment due to this brilliant piece of writing.
I’ll immediately seize your rss feed as I can’t find your emailsubscription hyperlink or e-newsletter service. Do you have any?Please allow me understand so that I could subscribe. Thanks.
Purdentix
I value the blog article.Really thank you! Really Great.
This is one awesome post. Keep writing.
This is one awesome article post. Keep writing.
Thanks a ton for finding the time to line all of this out for us. This kind of posting has been quite helpful if you ask me.
Im obliged for the article post.Really looking forward to read more. Fantastic.
Wow, great blog article.Much thanks again. Want more.
Awesome article post.Thanks Again. Fantastic.
Fastidious respond in return of this query with genuine arguments and explaining all concerning that.
Really enjoyed this post.Really thank you!
Im obliged for the post.Really thank you! Really Cool.
Im obliged for the post.Really looking forward to read more. Want more.
Very informative blog article.Much thanks again. Will read on…
I think this is a real great blog post.Much thanks again. Want more.
Thank you ever so for you blog post.Really thank you!
Enjoyed every bit of your article post.Really thank you! Much obliged.
Thank you ever so for you post.Really thank you! Keep writing.
Im obliged for the article post.Much thanks again. Keep writing.
Im obliged for the blog article.Really looking forward to read more. Will read on…
I cannot thank you enough for the blog article.Really thank you! Will read on…
Thank you for your post.Really thank you! Will read on…
I am so grateful for your article. Really Cool.
Really enjoyed this article post.Really looking forward to read more. Great.
I truly appreciate this blog article. Keep writing.
Im thankful for the blog post.Thanks Again. Great.
Great, thanks for sharing this article post.Really thank you! Much obliged.
Highly descriptive blog, I enjoyed that a lot.Will there be a part 2?
Im obliged for the post.Thanks Again. Awesome.
Muchos Gracias for your blog post. Keep writing.
I appreciate you sharing this blog post. Really Great.
Appreciate you sharing, great article post.Really looking forward to read more. Much obliged.
Wow, great blog article. Really Great.
This is one awesome article.Much thanks again. Cool.
Really informative post.Much thanks again. Want more.
I truly appreciate this article post.Really thank you!
Great, thanks for sharing this article post.Much thanks again. Awesome.
PINK SALT TRICK FOR WEIGHT LOSS
Im grateful for the blog article. Much obliged.
Very informative post.Much thanks again. Great.
Enjoyed every bit of your post. Really Great.
I am so grateful for your post.Thanks Again. Will read on…
Im obliged for the blog.Much thanks again. Really Great.
Thanks-a-mundo for the blog post. Will read on…
Very good article post.Thanks Again. Want more.
A big thank you for your blog article.Thanks Again. Keep writing.
Thanks so much for the blog post.Really looking forward to read more. Much obliged.
Looking forward to reading more. Great post.Thanks Again. Keep writing.
Thanks-a-mundo for the article.Really thank you! Really Cool.
I cannot thank you enough for the article.Really looking forward to read more. Much obliged.
I appreciate you sharing this article.Thanks Again. Keep writing.
Enjoyed every bit of your blog article.Really thank you! Really Cool.
Your style is very unique compared to other people I have read stuff from. Thanks for posting when you’ve got the opportunity, Guess I will just book mark this blog.
I am so grateful for your post. Will read on…
Great, thanks for sharing this post.Much thanks again. Want more.
Im thankful for the blog.Really thank you!
Really appreciate you sharing this post.Much thanks again. Really Great.
Really appreciate you sharing this blog post. Really Cool.
I appreciate you sharing this article.Much thanks again. Keep writing.
Thanks a lot for the blog article.Thanks Again. Really Cool.
Thanks a lot for the blog.Really thank you! Really Great.
Wow, great article post.Thanks Again. Fantastic.
Really informative blog article. Want more.
Thanks-a-mundo for the article.Really thank you! Keep writing.
I really enjoy the article. Much obliged.
I really liked your article post.Thanks Again.
Major thanks for the article.Really thank you!
Im thankful for the blog article.Really thank you! Really Great.
Very good post. Want more.
Really informative blog.Thanks Again. Great.
Very neat article.Really thank you!
Hey there! I’m at work surfing around your blog from my new apple iphone!Just wanted to say I love reading your blog and look forward to all yourposts! Keep up the superb work!
I value the article post.Really looking forward to read more. Cool.
I really liked your blog post.Really looking forward to read more. Will read on…
I truly appreciate this article post.Much thanks again. Great.
Muchos Gracias for your blog. Will read on…
Fantastic article.Thanks Again. Fantastic.
Muchos Gracias for your article post.Much thanks again. Awesome.
I really like and appreciate your post.Really thank you! Want more.
Appreciate you sharing, great blog post.Really thank you! Really Cool.
wow, awesome article. Fantastic.
Really informative post.Really thank you! Fantastic.
Thanks for the article post.Really looking forward to read more. Really Cool.
Muchos Gracias for your article. Much obliged.
A fascinating discussion is worth comment. I think that you need to publish more on this topic, it might not be a taboo subject but usually folks don’t discuss these issues. To the next! Cheers!!
Looking forward to reading more. Great article. Want more.
Thanks for sharing, this is a fantastic blog post. Great.
Hi there, constantly i used to check blog posts here early in the morning, because i enjoy to learn more and more.
Thank you ever so for you blog article.Really thank you! Really Great.
I am not certain where you’re getting your info, but good topic. I needs to spend some time studying more or figuring out more. Thank you for wonderful info I was in search of this information for my mission.
Really informative article post.Really thank you! Really Great.
Wow, great article.Much thanks again. Fantastic.
I think this is a real great blog.Really looking forward to read more. Really Great.
I really enjoy the article post.Thanks Again. Will read on…
Thanks so much for the blog article.Thanks Again. Really Cool.
There’s certainly a lot to know about this topic. I really like all the points you made.
I think this is a real great blog.Really thank you! Much obliged.
Great, thanks for sharing this blog post.Really thank you!
I really like and appreciate your post. Keep writing.
Looking forward to reading more. Great blog.Much thanks again. Want more.
Very informative blog post.Thanks Again. Really Cool.
Wow, great post.Much thanks again. Really Cool.Loading…
Your method of describing the whole thing in this paragraph is genuinely nice,all be capable of effortlessly understand it, Thanks a lot.
Do you have any video of that? I’d love to findout more details.
These are genuinely enormous ideas in concerning blogging. You have touched some nice things here. Any way keep up wrinting.
I loved your article.Much thanks again. Cool.
Thanks for one’s marvelous posting! I genuinely enjoyed reading it, you can be a great author.Iwill be sure to bookmark your blog and will come back later on.I want to encourage you to continue your great work,have a nice weekend!
Remarkable! Its really awesome paragraph, I have got much clear ideaon the topic of from this post.
wow, awesome article. Much obliged.
Ulasan yang terperinci membuat saya semakin paham topik yang dibahas. Saya merasa sangat dibantu dengan isi yang Anda sajikan ini.
That is really attention-grabbing, You’re an overly skilled blogger. I have joined your feed and stay up for in the hunt for more of your magnificent post. Also, I’ve shared your website in my social networks!
Really enjoyed this post.Thanks Again. Awesome.
Thank you ever so for you article post.Really thank you! Will read on…
Very good blog. Fantastic.
Hello.This post was really remarkable, especially since I was searching for thoughts on this topic last couple of days.
Thank you for your blog post.Really looking forward to read more.
A fascinating discussion is worth comment. There’s no doubt that that you ought to write more on this topic, it might not be a taboo subject but usually people don’t speak about such issues. To the next! Cheers!!
Thanks a lot for the post.Thanks Again. Great.
Very neat blog.Thanks Again.
A round of applause for your blog article.Much thanks again. Really Cool.
Really enjoyed this blog article.Much thanks again. Will read on…
Great, thanks for sharing this article post.Really thank you! Will read on…
Hey, thanks for the blog. Cool.
Great, thanks for sharing this article. Cool.
Fantastic article. Really Cool.
Say, you got a nice blog article. Much obliged.
Muchos Gracias for your article.Really thank you! Want more.
Thanks again for the blog article.Really looking forward to read more. Want more.
Say, you got a nice article post.Really looking forward to read more. Much obliged.
Thanks a lot for the post.Really thank you! Fantastic.
Appreciate you sharing, great blog article.Thanks Again. Will read on…
Thank you ever so for you blog post.Thanks Again. Awesome.
Thanks again for the post.Much thanks again.
Very neat blog article. Want more.
Great blog.Really thank you! Much obliged.
Fantastic blog. Much obliged.
Really enjoyed this blog.Really looking forward to read more. Really Cool.
Appreciate you sharing, great article post.Really looking forward to read more. Cool.
Very neat blog post.Really looking forward to read more. Much obliged.
I appreciate you sharing this blog article.Thanks Again. Much obliged.
I loved your article.Really thank you! Keep writing.
Thanks-a-mundo for the blog post.Thanks Again. Awesome.
Great blog article.Thanks Again. Really Cool.
I truly appreciate this article post.Thanks Again. Fantastic.
wow, awesome blog. Really Cool.
Awesome article post. Cool.
Im thankful for the post.Really thank you! Great.
I think this is a real great article.Really thank you! Much obliged.
Awesome blog post.Really thank you! Keep writing.
Awesome article post.Really thank you! Want more.
Thanks so much for the post. Awesome.
It’s hard to come by knowledgeable people for thistopic, but you sound like you know what you’re talking about!Thanks
Really appreciate you sharing this article post.Much thanks again. Cool.
I appreciate you sharing this post. Will read on…
I truly appreciate this blog post.Really looking forward to read more. Fantastic.
I really like and appreciate your blog post.Thanks Again. Want more.
Im grateful for the blog.Really looking forward to read more. Fantastic.
Fantastic article.Really thank you!
Really appreciate you sharing this blog post. Really Cool.
Muchos Gracias for your post.Much thanks again. Keep writing.
I value the blog post.Much thanks again. Really Great.
Great, thanks for sharing this article post.
wow, awesome blog post. Great.
Very good blog post. Really Great.
I value the blog article.Thanks Again. Will read on…
Say, you got a nice blog. Fantastic.
Very good blog. Will read on…
A round of applause for your blog article. Want more.
Muchos Gracias for your post.Much thanks again. Want more.
Really enjoyed this post.Really thank you! Fantastic.
I really enjoy the blog post.Really looking forward to read more.
A round of applause for your blog post.Much thanks again. Really Cool.
I loved your article.Much thanks again. Much obliged.
A round of applause for your blog.Much thanks again. Will read on…
I really like and appreciate your blog post.Really looking forward to read more. Really Cool.
Enjoyed every bit of your blog article.Really looking forward to read more. Great.
I really enjoy the article post.Really thank you! Much obliged.
Very good article.Really looking forward to read more. Really Great.
I really liked your blog.Much thanks again. Will read on…
I appreciate you sharing this article post.Really thank you! Much obliged.
I really like and appreciate your blog.Much thanks again. Much obliged.
Wow, great post.Really looking forward to read more. Fantastic.
I loved your blog post.Much thanks again.
Im grateful for the article.Thanks Again. Will read on…
Hey, thanks for the article post. Awesome.
Thanks again for the article post. Really Great.
I quite like reading through a post that can make people think. Also, thank you for permitting me to comment!
Very neat blog.Really looking forward to read more. Much obliged.
Major thanks for the blog post.Really thank you! Much obliged.
Thanks for sharing, this is a fantastic blog article.Really thank you! Really Great.
canadian pharmacy ltd online pharmacy china – express pharmacyus online pharmacy
Major thankies for the blog article.Really looking forward to read more. Will read on…
canadian pharmacy online ship to usa dextroamphetamine online pharmacy
how to get ivermectin in canada ivermectin topical
Hi there, just became aware of your blog through Google, and found that it’s really informative. I’m going to watch out for brussels. I’ll be grateful if you continue this in future. A lot of people will be benefited from your writing. Cheers!
This info is worth everyone’s attention. When can I find out more?0mniartist asmr
tizanidine hcl zanaflex for fibromyalgia tizanidine
Heklo there, just became aware of your blog through Google,and found that it is truly informative. I am going towatch out for brussels. I will appreciate if you continue this in future.Lots of people will be benefited from your writing.Cheers!
Wow, great article post.Really looking forward to read more. Keep writing.
Appreciate you sharing, great blog. Much obliged.
Thanks for the article.Much thanks again. Awesome.
Appreciate you sharing, great post.Thanks Again.
Say, you got a nice blog post.Thanks Again. Much obliged.
I really enjoy the article.Much thanks again. Great.
Hey, thanks for the article post.Much thanks again. Want more.
I really enjoy the blog post.Really looking forward to read more. Much obliged.
Hey, thanks for the blog post. Fantastic.
Hi there colleagues, its enormous piece of writing aboutteachingand fully defined, keep it up all the time.
Awesome blog.Much thanks again. Really Cool.
It’s genuinely very complex in this full of activity life tolisten news on TV, therefore I just use internetfor that purpose, and obtain the most recentinformation.
Thank you, I have recently been searching for info about this topic fora while and yours is the best I have discovered till now. But, what in regards to the bottom line?Are you certain concerning the source?
Great article.Really looking forward to read more. Much obliged.
magnificent points altogether, you simply received abrand new reader. What could you recommend in regards to your submit that you simply madesome days in the past? Any sure?
I haven’t checked in here for a while since I thought it was getting boring, but the last several posts are good quality so I guess I’ll add you back to my daily bloglist. You deserve it my friend 🙂
Excellent read, I just passed this onto a colleague who was doing a little research on that. And he actually bought me lunch because I found it for him smile Thus let me rephrase that: Thank you for lunch!
A round of applause for your blog post.Thanks Again. Fantastic.
A round of applause for your blog post.Really thank you! Much obliged.
I truly appreciate this post.Really thank you! Fantastic.
I am so grateful for your article. Really Cool.
It¦s actually a great and helpful piece of info. I am satisfied that you simply shared this useful info with us. Please keep us up to date like this. Thanks for sharing.
Great article.Much thanks again. Much obliged.
Im grateful for the article post.Much thanks again.
Really appreciate you sharing this blog. Awesome.
Hey, thanks for the article.Thanks Again. Really Cool.
Very informative post.Much thanks again. Want more.
Major thanks for the post.Much thanks again. Fantastic.
I cannot thank you enough for the post.Much thanks again. Really Cool.
Major thankies for the blog post.Really looking forward to read more. Want more.
Very good article post.Really thank you! Really Great.
I truly appreciate this article.Thanks Again. Great.
Really enjoyed this blog.Really thank you! Really Cool.
Fantastic article post.Thanks Again. Great.
Great article post.Thanks Again. Really Great.
Im thankful for the blog post. Fantastic.
I loved your blog post.Really looking forward to read more.
Enjoyed every bit of your blog post.Thanks Again.
Wow, great blog post.Really looking forward to read more. Want more.
Looking forward to reading more. Great article.Thanks Again. Keep writing.
I cannot thank you enough for the post.Thanks Again. Awesome.
Thank you for your blog post.Thanks Again. Keep writing.
I value the article post.Really looking forward to read more. Want more.
Appreciate you sharing, great blog article.Really looking forward to read more. Cool.
Muchos Gracias for your blog post.Much thanks again. Great.
Very good post.Really thank you! Cool.
Hey, thanks for the blog post.Really thank you! Awesome.
I really like and appreciate your post.Really thank you! Awesome.
I loved your post.Really thank you! Will read on…
Im obliged for the blog post.Really thank you! Much obliged.
Thanks for the post.Thanks Again.
Muchos Gracias for your blog. Really Cool.
Great blog article.Really thank you! Really Cool.
Looking forward to reading more. Great article post.Much thanks again. Fantastic.
Major thankies for the article post.Really thank you! Fantastic.
Really appreciate you sharing this blog article. Really Great.
I always was interested in this subject and still am, appreciate itfor putting up.my blog … Alpha Extracts Review
Hi there, of course this article is actually fastidious and I have learned lot of things from it concerning blogging. thanks.
Thanks for the auspicious writeup. It in reality used to be a entertainment account it.Look advanced to more introduced agreeable from you!By the way, how could we communicate?My blog post :: 그가 나의 잔디에 앉아
Great information. Lucky me I discovered your blog by chance (stumbleupon). I’ve bookmarked it for later!
Thanks again for the blog article.Thanks Again. Fantastic.
Wow, great blog article.Really looking forward to read more. Much obliged.
I really enjoy the blog article.Much thanks again. Fantastic.
Really informative blog.Really thank you! Great.
Great article post.Thanks Again. Really Cool.
Thanks a lot for the blog article.Much thanks again. Really Cool.
Its superb as your other content : D, thankyou for putting up. “History is a pact between the dead, the living, and the yet unborn.” by Edmund Burke.
Awesome article post.Really thank you! Really Great.
Hey, you used to write excellent, but the last few posts have been kinda boring¡K I miss your great writings. Past few posts are just a little bit out of track! come on!
Really enjoyed this blog article.Thanks Again. Want more.
side effects of vardenafil – how to use vardenafil pharmacy online order vardenafil men
Great, thanks for sharing this article post.Thanks Again. Want more.
PINK SALT TRICK RECIPE
PINK SALT TRICK FOR WEIGHT LOSS
Thanks so much for the blog post.Really thank you! Fantastic.
EREFORCE REVIEWS
EREFORCE REVIEWS
Thanks so much for the blog post.Really thank you! Will read on…
A round of applause for your blog post. Keep writing.
I appreciate you sharing this blog post.Really looking forward to read more. Keep writing.
I loved your blog article. Cool.
Enjoyed every bit of your article post.Really thank you! Want more.
Really informative article.Really looking forward to read more. Keep writing.
I loved your blog.Really looking forward to read more. Will read on…
Muchos Gracias for your blog article.Thanks Again.
Thanks for sharing, this is a fantastic blog.Really thank you! Great.
Fantastic article post. Keep writing.
Great post.Really looking forward to read more. Much obliged.
Thank you for your blog.Much thanks again. Much obliged.
augmentin 500 bactrim medline cephalexin gonnorhea
Im grateful for the blog article.Much thanks again. Great.
I appreciate you sharing this blog post. Really Great.
I really like and appreciate your blog article.Thanks Again.
I really enjoy the blog post. Awesome.
Very good blog article.Thanks Again. Cool.
WOW just what I was looking for. Came here by searching for windowunit
Im thankful for the blog.Really looking forward to read more. Awesome.
Thanks again for the blog.Much thanks again. Cool.
plaquenil anddogs plaquenil for osteoarthritis does plaquenil cause weight gain how plaquenil
Very interesting details you have noted , thanks for putting up. “The only thing worse than a man you can’t control is a man you can.” by Margo Kaufman.
Awesome article post.Thanks Again. Will read on…
Thanks for finally writing about > Πανελλήνιοπρωτάθλημα ποδηλασίας.– Cyclingworld.gr
You have brought up a very wonderful points,thank you for the post.my blog post – cyclical ketogenic
I am so grateful for your blog post.Really looking forward to read more. Keep writing.
I think this is a real great blog post.Really thank you! Much obliged.
Thanks so much for the article.Really thank you! Really Great.
Thanks again for the article. Keep writing.
Great blog post.Thanks Again. Want more.
Fantastic article post.Thanks Again. Really Cool.
Really appreciate you sharing this article. Much obliged.
Hey, thanks for the blog.Really thank you! Great.
I appreciate you sharing this blog post.Really looking forward to read more. Will read on…
Great, thanks for sharing this blog.Really thank you! Really Cool.
Thanks so much for the post.Really thank you! Want more.
I really like and appreciate your blog post.Much thanks again. Keep writing.
Very informative blog post. Really Cool.
Looking forward to reading more. Great article.Really thank you! Want more.
Great article.Thanks Again. Keep writing.
Very neat post.Thanks Again. Cool.
I appreciate you sharing this blog post. Want more.
I value the article post.Thanks Again. Really Great.
Very good blog.Thanks Again. Keep writing.
Awesome article.Thanks Again. Fantastic.
Thanks again for the blog.Really looking forward to read more. Fantastic.
Appreciate you sharing, great blog post.Thanks Again. Great.
I loved your blog.
Major thanks for the article.Really looking forward to read more. Fantastic.
I think this is a real great blog article.Much thanks again. Will read on…
Thanks again for the post.Really thank you! Keep writing.
Hi, after reading this remarkable article i am too glad to share my familiarity here with friends.
Very good article post.Really thank you! Will read on…
Looking forward to reading more. Great post.Really looking forward to read more. Really Great.
I really enjoy the article. Will read on…
Hey, thanks for the article post.Really looking forward to read more.
Im obliged for the blog article. Awesome.
Thank you ever so for you article.Much thanks again. Fantastic.
Great article.Thanks Again. Great.
Thanks for sharing, this is a fantastic blog.Much thanks again. Much obliged.
Wow, great blog post.Really looking forward to read more. Awesome.
Im grateful for the post.Much thanks again. Cool.
Wow, great blog post.Really thank you! Keep writing.
I appreciate you sharing this blog article.Thanks Again. Great.
Thank you ever so for you blog article.Thanks Again. Will read on…
Awesome post.Much thanks again. Really Cool.
Im thankful for the blog.Thanks Again. Want more.
Im grateful for the blog post.Really thank you! Great.
Im obliged for the blog post.Thanks Again. Really Great.
Im thankful for the article.Much thanks again. Great.
Major thanks for the post. Cool.
Very good post! We are linking to this particularly great article on our site. Keep up the great writing.
Greetings! Very useful advice in this particular article! It is the little changes that make the largest changes. Thanks a lot for sharing!
This website was… how do you say it? Relevant!! Finally I’ve found something which helped me. Appreciate it!
Can I simply say what a relief to find somebody who really understands what they’re discussing over the internet. You definitely know how to bring an issue to light and make it important. More and more people need to read this and understand this side of the story. I was surprised you aren’t more popular since you certainly have the gift.
The next time I read a blog, I hope that it does not disappoint me as much as this particular one. After all, Yes, it was my choice to read, however I actually thought you would have something useful to talk about. All I hear is a bunch of whining about something you could fix if you were not too busy looking for attention.
I love it whenever people get together and share ideas. Great blog, keep it up!
Hey there! I simply would like to offer you a huge thumbs up for your great info you have got here on this post. I’ll be returning to your web site for more soon.
Good post. I am going through many of these issues as well..
I was able to find good information from your articles.
It’s difficult to find well-informed people on this subject, however, you seem like you know what you’re talking about! Thanks
Aw, this was an extremely good post. Spending some time and actual effort to generate a great article… but what can I say… I put things off a whole lot and don’t manage to get nearly anything done.
Good write-up. I certainly appreciate this website. Stick with it!
Good write-up. I certainly appreciate this website. Thanks!
I love it when individuals get together and share views. Great website, continue the good work.
what does hydroxychloroquine treat chloroquine phosphate tablets
I used to be able to find good info from your blog articles.
Greetings! Very helpful advice within this article! It’s the little changes that produce the most important changes. Many thanks for sharing!
You’re so interesting! I do not suppose I have read through a single thing like that before. So nice to find another person with a few genuine thoughts on this issue. Really.. thanks for starting this up. This website is one thing that is required on the web, someone with a bit of originality.
May I simply just say what a relief to discover somebody who really understands what they are talking about on the net. You actually understand how to bring a problem to light and make it important. A lot more people need to look at this and understand this side of the story. It’s surprising you are not more popular since you certainly have the gift.
I was more than happy to uncover this website. I wanted to thank you for your time for this particularly fantastic read!! I definitely appreciated every bit of it and I have you bookmarked to check out new things in your blog.
Good article and straight to the point. I don’t know if this is really the best place to ask but do you people have any ideea where to hire some professional writers? Thanks in advance 🙂
Hi friends, pleasant article and pleasant urging commented here, I am really enjoying bythese.
Very good blog post. I certainly appreciate this website. Stick with it!
Hi there, I do think your site could possibly be having browser compatibility issues. When I take a look at your blog in Safari, it looks fine however when opening in I.E., it’s got some overlapping issues. I just wanted to give you a quick heads up! Besides that, fantastic blog!
I’m impressed, I have to admit. Rarely do I encounter a blog that’s equally educative and engaging, and without a doubt, you have hit the nail on the head. The issue is something which not enough people are speaking intelligently about. Now i’m very happy I found this during my search for something concerning this.
Hello! I could have sworn I’ve visited this blog before but after going through many of the articles I realized it’s new to me. Anyhow, I’m definitely delighted I found it and I’ll be book-marking it and checking back often!
I really like reading a post that can make people think. Also, many thanks for permitting me to comment.
This site was… how do I say it? Relevant!! Finally I have found something which helped me. Thank you!
I need to to thank you for this excellent read!! I absolutely loved every little bit of it. I have you bookmarked to check out new stuff you post…
This page definitely has all of the information I wanted concerning this subject and didn’t know who to ask.
There is definately a great deal to learn about this subject. I love all of the points you have made.
This page definitely has all the information and facts I wanted concerning this subject and didn’t know who to ask.
I could not resist commenting. Perfectly written.
I really like it whenever people come together and share ideas. Great website, keep it up!
Excellent article. I’m facing many of these issues as well..
This is a topic that’s close to my heart… Take care! Exactly where can I find the contact details for questions?
It’s difficult to find educated people for this topic, however, you seem like you know what you’re talking about! Thanks
This excellent website really has all of the information I needed about this subject and didn’t know who to ask.
Spot on with this write-up, I seriously feel this site needs a lot more attention. I’ll probably be back again to read through more, thanks for the advice.
Greetings! Very useful advice within this article! It is the little changes that make the biggest changes. Many thanks for sharing!
I want to to thank you for this good read!! I absolutely loved every little bit of it. I have got you book-marked to check out new stuff you post…
An intriguing discussion is definitely worth comment. I do think that you ought to publish more on this topic, it might not be a taboo subject but typically people don’t discuss these topics. To the next! Best wishes.
Pretty! This has been a really wonderful article. Thanks for providing these details.
Hi there! This blog post could not be written any better! Going through this post reminds me of my previous roommate! He continually kept preaching about this. I’ll send this article to him. Pretty sure he’s going to have a good read. I appreciate you for sharing!
This website really has all of the information I wanted concerning this subject and didn’t know who to ask.
Excellent post! We are linking to this particularly great content on our website. Keep up the good writing.
I’m impressed, I must say. Rarely do I come across a blog that’s equally educative and interesting, and let me tell you, you have hit the nail on the head. The problem is something that too few people are speaking intelligently about. I am very happy that I found this during my search for something regarding this.
This site was… how do you say it? Relevant!! Finally I have found something that helped me. Kudos.
Howdy! This blog post could not be written much better! Going through this post reminds me of my previous roommate! He always kept talking about this. I’ll send this post to him. Fairly certain he will have a great read. I appreciate you for sharing!
I’m impressed, I must say. Seldom do I come across a blog that’s both equally educative and interesting, and let me tell you, you’ve hit the nail on the head. The issue is an issue that too few men and women are speaking intelligently about. I’m very happy I came across this during my hunt for something relating to this.
Nice post. I learn something new and challenging on blogs I stumbleupon everyday. It will always be useful to read through content from other writers and use something from other web sites.
Hi, I think your website could possibly be having web browser compatibility problems. Whenever I take a look at your website in Safari, it looks fine but when opening in I.E., it’s got some overlapping issues. I simply wanted to give you a quick heads up! Besides that, excellent website!
You should take part in a contest for one of the best blogs on the web. I most certainly will highly recommend this site!
I could not refrain from commenting. Very well written.
I could not refrain from commenting. Well written.
I could not refrain from commenting. Very well written!
This site was… how do you say it? Relevant!! Finally I have found something that helped me. Many thanks.
I blog quite often and I seriously thank you for your information. This article has truly peaked my interest. I’m going to book mark your blog and keep checking for new details about once a week. I subscribed to your Feed as well.
Pretty! This was an extremely wonderful article. Thanks for providing this info.
Right here is the perfect website for anyone who wishes to understand this topic. You know so much its almost hard to argue with you (not that I personally will need to…HaHa). You definitely put a fresh spin on a subject that has been written about for decades. Excellent stuff, just excellent.
Good site you have got here.. It’s difficult to find high-quality writing like yours these days. I truly appreciate people like you! Take care!!
After exploring a number of the articles on your web page, I truly appreciate your technique of blogging. I saved it to my bookmark site list and will be checking back soon. Please check out my web site as well and tell me how you feel.
Hello there! I simply wish to give you a big thumbs up for your excellent info you have here on this post. I am coming back to your web site for more soon.
I couldn’t resist commenting. Perfectly written!
Hi there, I do believe your site could be having web browser compatibility issues. Whenever I look at your blog in Safari, it looks fine however, if opening in I.E., it’s got some overlapping issues. I merely wanted to provide you with a quick heads up! Apart from that, excellent site.
Can I simply just say what a comfort to uncover somebody who really understands what they’re talking about on the net. You certainly know how to bring a problem to light and make it important. More and more people must read this and understand this side of the story. It’s surprising you’re not more popular since you most certainly possess the gift.
I love it when individuals come together and share ideas. Great site, continue the good work.
I used to be able to find good information from your blog articles.
Howdy! I simply want to offer you a huge thumbs up for the excellent info you’ve got right here on this post. I will be returning to your web site for more soon.
Excellent article! We are linking to this particularly great content on our website. Keep up the good writing.
I’m very pleased to uncover this site. I want to to thank you for ones time for this fantastic read!! I definitely savored every little bit of it and I have you bookmarked to check out new things in your blog.
I’m very pleased to find this web site. I need to to thank you for ones time for this particularly wonderful read!! I definitely appreciated every part of it and i also have you book-marked to check out new information on your site.
Your style is unique compared to other folks I have read stuff from. I appreciate you for posting when you’ve got the opportunity, Guess I’ll just bookmark this page.
Can I simply just say what a comfort to uncover someone that actually understands what they’re discussing on the net. You definitely understand how to bring a problem to light and make it important. More people must look at this and understand this side of the story. I can’t believe you aren’t more popular since you most certainly possess the gift.
It’s hard to come by well-informed people in this particular subject, however, you seem like you know what you’re talking about! Thanks
It is in point of fact a nice and useful piece of information. I am glad that you shared this helpful info with us. Please stay us informed like this. Thank you for sharing.
Very good write-up. I certainly love this site. Stick with it!
Way cool! Some very valid points! I appreciate you writing this article and the rest of the site is extremely good.
I absolutely love your website.. Very nice colors & theme. Did you develop this amazing site yourself? Please reply back as I’m planning to create my own blog and want to learn where you got this from or what the theme is called. Appreciate it!
You have made some really good points there. I checked on the web to find out more about the issue and found most people will go along with your views on this site.
Having read this I thought it was very informative. I appreciate you taking the time and effort to put this article together. I once again find myself personally spending a lot of time both reading and posting comments. But so what, it was still worthwhile!
I couldn’t resist commenting. Well written.
I really like it whenever people get together and share ideas. Great site, continue the good work!
Having read this I thought it was extremely enlightening. I appreciate you finding the time and energy to put this information together. I once again find myself personally spending a significant amount of time both reading and leaving comments. But so what, it was still worth it!
There is certainly a lot to find out about this topic. I really like all of the points you made.
Can I simply just say what a comfort to discover an individual who actually understands what they’re talking about on the net. You certainly know how to bring an issue to light and make it important. More and more people really need to check this out and understand this side of the story. I was surprised that you’re not more popular since you certainly possess the gift.
I’m impressed, I have to admit. Seldom do I encounter a blog that’s equally educative and entertaining, and without a doubt, you’ve hit the nail on the head. The issue is an issue that not enough people are speaking intelligently about. I’m very happy I found this in my hunt for something concerning this.
I was able to find good advice from your articles.
After I originally left a comment I seem to have clicked the -Notify me when new comments are added- checkbox and from now on whenever a comment is added I recieve four emails with the exact same comment. Is there an easy method you are able to remove me from that service? Appreciate it.
I used to be able to find good advice from your blog articles.
Hi! I simply would like to give you a huge thumbs up for your great info you’ve got right here on this post. I am coming back to your site for more soon.
Way cool! Some very valid points! I appreciate you penning this article plus the rest of the website is extremely good.
This is a good tip particularly to those fresh to the blogosphere. Brief but very precise information… Appreciate your sharing this one. A must read post.
This website was… how do I say it? Relevant!! Finally I have found something that helped me. Many thanks.
It’s difficult to find educated people for this topic, however, you seem like you know what you’re talking about! Thanks
Wonderful post! We are linking to this great article on our website. Keep up the great writing.
Everything is very open with a precise clarification of the challenges. It was really informative. Your site is useful. Thanks for sharing.
Everyone loves it whenever people come together and share ideas. Great website, stick with it!
I’m amazed, I must say. Seldom do I encounter a blog that’s equally educative and engaging, and let me tell you, you have hit the nail on the head. The problem is something that not enough people are speaking intelligently about. I’m very happy I came across this in my hunt for something concerning this.
I will right away grasp your rss as I can’t find your email subscription link or e-newsletter service. Do you’ve any? Kindly let me recognise so that I may subscribe. Thanks.
Great post. I was checking constantly this blog and I’m inspired! Very helpful information specifically the closing phase 🙂 I handle such info a lot. I was seeking this particular information for a very long time. Thank you and good luck.
An outstanding share! I’ve just forwarded this onto a colleague who had been doing a little homework on this. And he actually bought me breakfast because I discovered it for him… lol. So allow me to reword this…. Thanks for the meal!! But yeah, thanks for spending some time to discuss this issue here on your web page.
Saved as a favorite, I love your site.
You ought to be a part of a contest for one of the most useful blogs on the web. I’m going to recommend this site!
Way cool! Some very valid points! I appreciate you penning this article and the rest of the site is really good.
Can I just say what a relief to uncover someone that really knows what they’re talking about on the web. You actually know how to bring an issue to light and make it important. A lot more people must look at this and understand this side of the story. It’s surprising you’re not more popular since you certainly have the gift.
You’ve made some good points there. I checked on the internet for more information about the issue and found most people will go along with your views on this website.
An impressive share! I’ve just forwarded this onto a friend who has been doing a little homework on this. And he actually ordered me lunch due to the fact that I found it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanx for spending the time to discuss this subject here on your website.
A fascinating discussion is definitely worth comment. I do think that you should write more about this subject matter, it may not be a taboo subject but typically people don’t talk about these subjects. To the next! Kind regards.
Your style is really unique compared to other people I’ve read stuff from. Many thanks for posting when you’ve got the opportunity, Guess I will just book mark this page.
Having read this I thought it was rather informative. I appreciate you spending some time and effort to put this short article together. I once again find myself personally spending a significant amount of time both reading and leaving comments. But so what, it was still worth it!
Pretty! This has been an extremely wonderful post. Many thanks for supplying these details.
Pretty! This was an incredibly wonderful post. Thank you for supplying these details.
After looking at a few of the articles on your web page, I seriously appreciate your technique of blogging. I book marked it to my bookmark site list and will be checking back soon. Take a look at my website as well and let me know your opinion.
An intriguing discussion is worth comment. I believe that you ought to publish more about this subject, it might not be a taboo matter but generally folks don’t speak about these topics. To the next! Cheers.
Hi! I could have sworn I’ve been to your blog before but after looking at some of the articles I realized it’s new to me. Nonetheless, I’m definitely pleased I discovered it and I’ll be bookmarking it and checking back regularly!
This website really has all of the information I wanted about this subject and didn’t know who to ask.
I’d like to thank you for the efforts you’ve put in writing this website. I am hoping to see the same high-grade blog posts from you later on as well. In fact, your creative writing abilities has inspired me to get my very own site now 😉
Oh my goodness! Incredible article dude! Thanks, However I am having issues with your RSS. I don’t understand why I am unable to join it. Is there anybody else having similar RSS issues? Anyone that knows the answer can you kindly respond? Thanks!!
This site was… how do you say it? Relevant!! Finally I’ve found something which helped me. Cheers.
Good information. Lucky me I ran across your website by accident (stumbleupon). I have book-marked it for later.
I must thank you for the efforts you’ve put in writing this blog. I’m hoping to see the same high-grade content by you later on as well. In truth, your creative writing abilities has motivated me to get my own, personal website now 😉
This web site truly has all the information and facts I wanted concerning this subject and didn’t know who to ask.
I’m extremely pleased to uncover this great site. I wanted to thank you for ones time for this particularly wonderful read!! I definitely liked every bit of it and i also have you saved to fav to see new information on your blog.
Way cool! Some extremely valid points! I appreciate you writing this post and the rest of the site is also very good.
Aw, this was a really good post. Finding the time and actual effort to create a really good article… but what can I say… I procrastinate a lot and never seem to get anything done.
I’m amazed, I must say. Seldom do I come across a blog that’s both equally educative and interesting, and let me tell you, you have hit the nail on the head. The issue is something not enough people are speaking intelligently about. I am very happy I came across this in my search for something regarding this.
Hi! I could have sworn I’ve been to this blog before but after going through many of the posts I realized it’s new to me. Regardless, I’m certainly delighted I stumbled upon it and I’ll be bookmarking it and checking back often.
Next time I read a blog, I hope that it doesn’t disappoint me just as much as this particular one. After all, I know it was my choice to read, however I truly believed you’d have something helpful to say. All I hear is a bunch of complaining about something you can fix if you were not too busy looking for attention.
Hello there! I could have sworn I’ve visited this blog before but after going through many of the posts I realized it’s new to me. Anyhow, I’m certainly pleased I found it and I’ll be book-marking it and checking back frequently.
An outstanding share! I have just forwarded this onto a coworker who has been doing a little homework on this. And he actually ordered me breakfast due to the fact that I discovered it for him… lol. So allow me to reword this…. Thanks for the meal!! But yeah, thanx for spending some time to talk about this subject here on your site.
I truly love your site.. Very nice colors & theme. Did you develop this site yourself? Please reply back as I’m trying to create my very own website and would love to learn where you got this from or exactly what the theme is called. Cheers!
An impressive share! I’ve just forwarded this onto a colleague who was doing a little homework on this. And he actually bought me dinner due to the fact that I discovered it for him… lol. So allow me to reword this…. Thank YOU for the meal!! But yeah, thanx for spending time to discuss this issue here on your site.
Hello! I could have sworn I’ve been to this web site before but after browsing through some of the posts I realized it’s new to me. Anyhow, I’m definitely delighted I found it and I’ll be bookmarking it and checking back frequently.
That is a very good tip particularly to those new to the blogosphere. Short but very precise info… Thanks for sharing this one. A must read article.
I must thank you for the efforts you have put in writing this blog. I’m hoping to view the same high-grade content by you later on as well. In fact, your creative writing abilities has inspired me to get my own website now 😉
I need to to thank you for this fantastic read!! I certainly loved every bit of it. I’ve got you bookmarked to check out new things you post…
Greetings! Very helpful advice within this post! It is the little changes which will make the greatest changes. Thanks a lot for sharing!
Judge denies November request to remove Britney Spears’ father as her co-conservator..1
This is a topic that is close to my heart… Thank you! Where can I find the contact details for questions?
Pretty! This has been an incredibly wonderful post. Thank you for supplying these details.
An intriguing discussion is definitely worth comment. There’s no doubt that that you need to publish more on this subject, it might not be a taboo matter but usually people don’t discuss these subjects. To the next! Kind regards.
Great post! We will be linking to this great post on our website. Keep up the great writing.
Howdy! This article could not be written much better! Looking at this article reminds me of my previous roommate! He always kept talking about this. I most certainly will forward this information to him. Fairly certain he’ll have a good read. Many thanks for sharing!
I used to be able to find good information from your blog articles.
I used to be able to find good information from your articles.
Everything is very open with a really clear description of the issues. It was definitely informative. Your site is very useful. Thank you for sharing.
This is a topic that is close to my heart… Take care! Where can I find the contact details for questions?
This website definitely has all of the information I wanted about this subject and didn’t know who to ask.
Howdy, I do think your web site might be having web browser compatibility issues. When I take a look at your website in Safari, it looks fine however, when opening in Internet Explorer, it has some overlapping issues. I just wanted to provide you with a quick heads up! Aside from that, fantastic website!
Excellent web site you have got here.. It’s hard to find good quality writing like yours these days. I seriously appreciate individuals like you! Take care!!
Pretty! This was a really wonderful post. Thank you for supplying this information.
I’m amazed, I have to admit. Seldom do I encounter a blog that’s both equally educative and interesting, and let me tell you, you’ve hit the nail on the head. The problem is something that too few men and women are speaking intelligently about. Now i’m very happy that I found this in my search for something concerning this.
You need to take part in a contest for one of the highest quality websites online. I am going to recommend this blog!
Great article. I will be facing some of these issues as well..
You need to be a part of a contest for one of the most useful websites online. I will recommend this blog!
Everything is very open with a very clear clarification of the challenges. It was truly informative. Your website is extremely helpful. Thanks for sharing!
doktor kbb
dr kandulu
Very good post! We are linking to this particularly great article on our website. Keep up the great writing.
dr şaban
Having read this I believed it was very enlightening. I appreciate you taking the time and effort to put this short article together. I once again find myself spending a significant amount of time both reading and posting comments. But so what, it was still worthwhile.
Very good information. Lucky me I ran across your website by accident (stumbleupon). I have bookmarked it for later!
This excellent website truly has all the info I wanted about this subject and didn’t know who to ask.
I seriously love your site.. Excellent colors & theme. Did you create this web site yourself? Please reply back as I’m trying to create my own personal blog and would like to find out where you got this from or just what the theme is named. Kudos!
I was extremely pleased to uncover this great site. I wanted to thank you for ones time for this wonderful read!! I definitely enjoyed every bit of it and i also have you book-marked to look at new stuff in your website.
I used to be able to find good info from your blog posts.
Pretty! This has been a really wonderful post. Thanks for supplying this info.
Hi! I simply wish to offer you a big thumbs up for the great information you’ve got right here on this post. I’ll be coming back to your website for more soon.
I couldn’t resist commenting. Well written!
Good post. I will be experiencing a few of these issues as well..
Next time I read a blog, I hope that it won’t disappoint me just as much as this particular one. I mean, I know it was my choice to read, however I genuinely believed you would have something interesting to talk about. All I hear is a bunch of moaning about something you could fix if you weren’t too busy searching for attention.
This page certainly has all of the information and facts I wanted concerning this subject and didn’t know who to ask.
That is a great tip especially to those new to the blogosphere. Brief but very precise information… Thanks for sharing this one. A must read post!
You’ve made some really good points there. I checked on the web to find out more about the issue and found most people will go along with your views on this website.
This is a topic that is close to my heart… Cheers! Where are your contact details though?
Very good info. Lucky me I recently found your site by accident (stumbleupon). I’ve saved as a favorite for later!
Very nice blog post. I definitely appreciate this site. Thanks!
I was very happy to find this site. I wanted to thank you for ones time due to this wonderful read!! I definitely loved every bit of it and I have you saved to fav to look at new things on your web site.
Next time I read a blog, I hope that it doesn’t disappoint me just as much as this one. After all, Yes, it was my choice to read through, but I truly believed you would have something useful to say. All I hear is a bunch of complaining about something that you can fix if you were not too busy looking for attention.
Next time I read a blog, I hope that it does not disappoint me as much as this particular one. I mean, I know it was my choice to read through, nonetheless I genuinely believed you’d have something useful to say. All I hear is a bunch of complaining about something you could fix if you were not too busy seeking attention.
Everyone loves it whenever people come together and share ideas. Great site, continue the good work!
Howdy, I do believe your web site may be having web browser compatibility issues. When I take a look at your web site in Safari, it looks fine however, if opening in IE, it’s got some overlapping issues. I simply wanted to give you a quick heads up! Aside from that, great site.
bookmarked!!, I really like your blog.
Hello, I think your website could be having web browser compatibility issues. When I take a look at your web site in Safari, it looks fine however, when opening in IE, it’s got some overlapping issues. I just wanted to provide you with a quick heads up! Other than that, excellent blog!
Oh my goodness! Incredible article dude! Many thanks, However I am having issues with your RSS. I don’t understand the reason why I can’t join it. Is there anybody getting similar RSS issues? Anybody who knows the solution can you kindly respond? Thanx!
You’re so cool! I do not think I’ve truly read through a single thing like that before. So good to discover another person with some unique thoughts on this subject. Really.. many thanks for starting this up. This site is one thing that is required on the web, someone with a little originality.
esteworld
GELATIN TRICK
Excellent article. I’m going through a few of these issues as well..
Pretty! This was an incredibly wonderful article. Many thanks for supplying this info.
After I originally commented I seem to have clicked the -Notify me when new comments are added- checkbox and now whenever a comment is added I get 4 emails with the same comment. Is there a means you are able to remove me from that service? Thanks a lot.
I couldn’t refrain from commenting. Perfectly written!
Hello my friend! I wish to say that this post is amazing, nice written and comewith approximately all vital infos. I would like to peer more posts like this .
esteworld
Right here is the right webpage for everyone who wishes to find out about this topic. You know so much its almost tough to argue with you (not that I actually will need to…HaHa). You definitely put a new spin on a topic which has been discussed for ages. Great stuff, just wonderful.
There’s certainly a lot to know about this topic. I really like all of the points you have made.
After looking over a few of the blog articles on your site, I truly appreciate your way of writing a blog. I book-marked it to my bookmark webpage list and will be checking back soon. Please visit my website too and let me know what you think.
I’m excited to discover this site. I need to to thank you for ones time for this fantastic read!! I definitely savored every little bit of it and I have you book-marked to see new things in your site.
An outstanding share! I have just forwarded this onto a co-worker who had been doing a little homework on this. And he actually bought me breakfast simply because I discovered it for him… lol. So let me reword this…. Thanks for the meal!! But yeah, thanx for spending some time to discuss this topic here on your blog.
Aw, this was an exceptionally good post. Taking a few minutes and actual effort to produce a top notch article… but what can I say… I procrastinate a whole lot and don’t seem to get anything done.
I like reading a post that will make people think. Also, many thanks for permitting me to comment.
Howdy, I believe your blog might be having web browser compatibility problems. Whenever I take a look at your web site in Safari, it looks fine however when opening in I.E., it’s got some overlapping issues. I just wanted to provide you with a quick heads up! Besides that, wonderful website.
Spot on with this write-up, I actually believe this web site needs a great deal more attention. I’ll probably be returning to read more, thanks for the info.
This site truly has all the information I wanted concerning this subject and didn’t know who to ask.
This is the right website for anybody who really wants to find out about this topic. You know so much its almost tough to argue with you (not that I personally will need to…HaHa). You definitely put a new spin on a subject which has been discussed for decades. Excellent stuff, just excellent.
It’s difficult to find knowledgeable people for this subject, but you seem like you know what you’re talking about! Thanks
When I originally left a comment I seem to have clicked the -Notify me when new comments are added- checkbox and now every time a comment is added I receive four emails with the same comment. There has to be an easy method you are able to remove me from that service? Appreciate it.
This is a topic that is near to my heart… Thank you! Exactly where are your contact details though?
An intriguing discussion is definitely worth comment. I think that you need to publish more about this topic, it may not be a taboo matter but generally people don’t talk about such subjects. To the next! Best wishes.
Way cool! Some extremely valid points! I appreciate you writing this post plus the rest of the site is really good.
Next time I read a blog, Hopefully it won’t fail me as much as this one. After all, Yes, it was my choice to read through, but I truly believed you would have something interesting to say. All I hear is a bunch of whining about something you can fix if you weren’t too busy seeking attention.
When I originally commented I seem to have clicked the -Notify me when new comments are added- checkbox and from now on whenever a comment is added I receive 4 emails with the exact same comment. There has to be an easy method you can remove me from that service? Thanks.
Great article! We are linking to this great post on our website. Keep up the good writing.
Good article. I am experiencing some of these issues as well..
This blog was… how do you say it? Relevant!! Finally I’ve found something that helped me. Many thanks!
I wanted to thank you for this fantastic read!! I definitely loved every little bit of it. I have got you bookmarked to check out new things you post…
This site really has all the information I wanted concerning this subject and didn’t know who to ask.
Excellent article. I’m dealing with many of these issues as well..
You ought to take part in a contest for one of the best sites on the web. I’m going to highly recommend this site!
Saved as a favorite, I love your web site!
Very good post! We are linking to this great post on our website. Keep up the good writing.
Good article. I definitely appreciate this website. Keep writing!
sapphire hair clinic
medhair
Superb post however I was wondering if you could write a litte more on this subject? I’d be very grateful if you could elaborate a little bit more. Thank you!
Great post. I am going through many of these issues as well..
I was very pleased to uncover this page. I want to to thank you for your time just for this wonderful read!! I definitely loved every part of it and I have you book-marked to check out new information in your web site.
I absolutely love your blog.. Pleasant colors & theme. Did you create this amazing site yourself? Please reply back as I’m hoping to create my own personal blog and want to learn where you got this from or just what the theme is named. Many thanks!
I love it when folks get together and share ideas. Great blog, continue the good work.
Hi, I do think your website could be having internet browser compatibility problems. When I take a look at your site in Safari, it looks fine however, when opening in Internet Explorer, it’s got some overlapping issues. I simply wanted to provide you with a quick heads up! Apart from that, wonderful website.
After going over a handful of the articles on your site, I honestly like your way of writing a blog. I bookmarked it to my bookmark website list and will be checking back in the near future. Please visit my website too and let me know your opinion.
Pretty! This was an extremely wonderful article. Many thanks for providing this info.
I need to to thank you for this good read!! I certainly loved every little bit of it. I’ve got you saved as a favorite to look at new stuff you post…
This site really has all of the info I needed concerning this subject and didn’t know who to ask.
Everything is very open with a really clear clarification of the issues. It was definitely informative. Your website is useful. Many thanks for sharing!
A fascinating discussion is worth comment. There’s no doubt that that you need to write more on this subject, it might not be a taboo matter but typically folks don’t speak about such subjects. To the next! Many thanks.
Great article! We will be linking to this great article on our website. Keep up the great writing.
Pretty! This has been a really wonderful post. Many thanks for providing these details.
I’m impressed, I must say. Seldom do I come across a blog that’s both educative and interesting, and without a doubt, you have hit the nail on the head. The problem is something that not enough people are speaking intelligently about. Now i’m very happy I came across this in my hunt for something concerning this.
Great info. Lucky me I found your website by accident (stumbleupon). I’ve book-marked it for later.
An interesting discussion is worth comment. I do think that you need to write more about this issue, it may not be a taboo matter but usually people do not speak about these subjects. To the next! Cheers!
I’ve read some good stuff here. Definitely worth bookmarking for revisiting. I surprise how much effort you put to make such a great informative site.
I must thank you for the efforts you’ve put in penning this site. I am hoping to view the same high-grade blog posts by you later on as well. In fact, your creative writing abilities has motivated me to get my own blog now 😉
I’d like to thank you for the efforts you have put in writing this site. I am hoping to view the same high-grade content by you later on as well. In truth, your creative writing abilities has motivated me to get my own website now 😉
May I simply say what a comfort to uncover an individual who truly understands what they’re discussing online. You actually understand how to bring a problem to light and make it important. More people have to check this out and understand this side of the story. I can’t believe you aren’t more popular because you certainly possess the gift.
Hi there! I could have sworn I’ve visited this blog before but after going through a few of the posts I realized it’s new to me. Anyhow, I’m definitely pleased I found it and I’ll be book-marking it and checking back frequently!
Can I just say what a comfort to find a person that genuinely understands what they’re discussing on the internet. You certainly understand how to bring an issue to light and make it important. More people should look at this and understand this side of the story. I was surprised you’re not more popular because you definitely have the gift.
Good article. I’m experiencing many of these issues as well..
Spot on with this write-up, I truly feel this site needs a lot more attention. I’ll probably be returning to see more, thanks for the advice.
salt trick for men
I’m amazed, I have to admit. Seldom do I encounter a blog that’s both equally educative and entertaining, and without a doubt, you’ve hit the nail on the head. The issue is something too few folks are speaking intelligently about. I’m very happy I stumbled across this in my search for something concerning this.
After I initially left a comment I seem to have clicked the -Notify me when new comments are added- checkbox and now each time a comment is added I get four emails with the same comment. There has to be an easy method you can remove me from that service? Many thanks.
Greetings, There’s no doubt that your web site may be having web browser compatibility issues. Whenever I look at your site in Safari, it looks fine however when opening in IE, it’s got some overlapping issues. I just wanted to provide you with a quick heads up! Apart from that, wonderful blog.
When I originally left a comment I seem to have clicked on the -Notify me when new comments are added- checkbox and now each time a comment is added I recieve 4 emails with the exact same comment. There has to be a means you are able to remove me from that service? Thanks a lot.
Aw, this was an exceptionally nice post. Taking a few minutes and actual effort to create a very good article… but what can I say… I hesitate a whole lot and don’t seem to get anything done.
You have made some decent points there. I checked on the web to learn more about the issue and found most individuals will go along with your views on this website.
Spot on with this write-up, I absolutely think this web site needs a great deal more attention. I’ll probably be returning to read through more, thanks for the information.
Good article. I will be dealing with a few of these issues as well..
Howdy! This article could not be written much better! Reading through this post reminds me of my previous roommate! He continually kept talking about this. I am going to forward this information to him. Fairly certain he will have a good read. Many thanks for sharing!
I really love your site.. Very nice colors & theme. Did you create this site yourself? Please reply back as I’m looking to create my own personal site and would like to find out where you got this from or just what the theme is called. Kudos.
BARIATRIC GELATIN TRICK
Hey there! I just want to offer you a huge thumbs up for the excellent info you have here on this post. I’ll be coming back to your website for more soon.
GELATIN TRICK
Oh my goodness! Awesome article dude! Thanks, However I am experiencing difficulties with your RSS. I don’t understand the reason why I can’t subscribe to it. Is there anybody else getting identical RSS problems? Anybody who knows the answer can you kindly respond? Thanks!!
The very next time I read a blog, I hope that it doesn’t fail me just as much as this one. After all, I know it was my choice to read, nonetheless I genuinely believed you’d have something interesting to say. All I hear is a bunch of whining about something you could fix if you were not too busy looking for attention.
I’m amazed, I must say. Rarely do I encounter a blog that’s both educative and interesting, and let me tell you, you have hit the nail on the head. The issue is something that not enough folks are speaking intelligently about. I’m very happy that I found this in my search for something relating to this.
Good post. I learn something totally new and challenging on websites I stumbleupon every day. It’s always exciting to read content from other writers and use something from other web sites.
Great post. I will be dealing with many of these issues as well..
BARIATRIC GELATIN TRICK
Way cool! Some very valid points! I appreciate you penning this write-up plus the rest of the website is also very good.
Aw, this was an exceptionally good post. Finding the time and actual effort to generate a superb article… but what can I say… I put things off a whole lot and never manage to get anything done.
Aw, this was an exceptionally good post. Taking the time and actual effort to produce a great article… but what can I say… I hesitate a whole lot and never seem to get nearly anything done.
Your style is really unique in comparison to other people I’ve read stuff from. Thanks for posting when you’ve got the opportunity, Guess I will just book mark this page.
Very interesting points you have noted, appreciate it for posting. “The only thing worse than a man you can’t control is a man you can.” by Margo Kaufman.
You’ve made some good points there. I looked on the web to learn more about the issue and found most people will go along with your views on this web site.
I want to to thank you for this great read!! I absolutely enjoyed every little bit of it. I have got you book-marked to check out new things you post…
Howdy, I believe your web site could be having web browser compatibility problems. When I take a look at your blog in Safari, it looks fine however, if opening in I.E., it’s got some overlapping issues. I just wanted to give you a quick heads up! Aside from that, fantastic blog.
Your style is really unique in comparison to other people I have read stuff from. Thank you for posting when you’ve got the opportunity, Guess I’ll just bookmark this site.
Hello, I believe your website may be having internet browser compatibility problems. When I look at your web site in Safari, it looks fine however, when opening in IE, it has some overlapping issues. I simply wanted to provide you with a quick heads up! Apart from that, excellent site!
After looking into a handful of the blog posts on your web site, I truly appreciate your way of blogging. I bookmarked it to my bookmark site list and will be checking back soon. Take a look at my website too and let me know your opinion.
I needed to thank you for this great read!! I absolutely loved every bit of it. I’ve got you book-marked to look at new stuff you post…
Great information. Lucky me I discovered your blog by accident (stumbleupon). I have book marked it for later.
There’s certainly a great deal to know about this subject. I like all the points you made.
Spot on with this write-up, I truly believe this amazing site needs far more attention. I’ll probably be back again to see more, thanks for the information.
This is the perfect web site for everyone who really wants to find out about this topic. You understand so much its almost tough to argue with you (not that I personally would want to…HaHa). You certainly put a new spin on a subject that’s been discussed for years. Wonderful stuff, just excellent.
This web site truly has all of the info I wanted about this subject and didn’t know who to ask.
This is a topic which is close to my heart… Best wishes! Where can I find the contact details for questions?
Great post! We are linking to this great article on our website. Keep up the good writing.
Great article. I am facing a few of these issues as well..
Spot on with this write-up, I truly believe this amazing site needs much more attention. I’ll probably be returning to read more, thanks for the advice!
An impressive share! I have just forwarded this onto a friend who was doing a little research on this. And he actually bought me breakfast due to the fact that I stumbled upon it for him… lol. So allow me to reword this…. Thanks for the meal!! But yeah, thanks for spending some time to talk about this topic here on your blog.
That is a really good tip especially to those fresh to the blogosphere. Short but very precise information… Appreciate your sharing this one. A must read article!
The very next time I read a blog, I hope that it doesn’t fail me just as much as this particular one. After all, Yes, it was my choice to read through, but I genuinely thought you’d have something helpful to say. All I hear is a bunch of complaining about something you could fix if you were not too busy looking for attention.
Spot on with this write-up, I seriously believe this website needs a lot more attention. I’ll probably be returning to read more, thanks for the info!
MITOLYN
I’m amazed, I must say. Rarely do I come across a blog that’s equally educative and entertaining, and without a doubt, you have hit the nail on the head. The issue is an issue that not enough men and women are speaking intelligently about. Now i’m very happy that I found this in my hunt for something relating to this.
GELATIN TRICK
Way cool! Some extremely valid points! I appreciate you writing this post and also the rest of the site is really good.
I couldn’t resist commenting. Very well written.
I simply wanted to make a simple comment in order to express gratitude to you for all of the pleasant steps you are giving out on this website. My time consuming internet research has finally been rewarded with high-quality ideas to go over with my company. I ‘d repeat that we website visitors are unquestionably fortunate to dwell in a really good community with many special individuals with insightful hints. I feel really privileged to have seen your entire site and look forward to plenty of more thrilling times reading here. Thanks a lot again for all the details.
I’ve been surfing online more than three hours as of late, but I never discovered any fascinating article like yours. It?¦s beautiful value sufficient for me. In my view, if all web owners and bloggers made good content as you did, the web will probably be a lot more useful than ever before.
Having read this I thought it was extremely enlightening. I appreciate you spending some time and effort to put this information together. I once again find myself spending a significant amount of time both reading and posting comments. But so what, it was still worthwhile.
Howdy! This post could not be written much better! Looking through this article reminds me of my previous roommate! He continually kept preaching about this. I most certainly will send this post to him. Fairly certain he will have a great read. Many thanks for sharing!
It’s hard to come by knowledgeable people about this topic, however, you seem like you know what you’re talking about! Thanks
Howdy! I could have sworn I’ve visited this site before but after browsing through a few of the posts I realized it’s new to me. Anyways, I’m definitely pleased I found it and I’ll be book-marking it and checking back regularly.
Hi there! I just wish to offer you a big thumbs up for the great info you have got here on this post. I am returning to your web site for more soon.
Hi, I do think this is an excellent web site. I stumbledupon it 😉 I will come back yet again since i have saved as a favorite it. Money and freedom is the best way to change, may you be rich and continue to guide other people.
A fascinating discussion is definitely worth comment. I do believe that you ought to write more about this subject matter, it might not be a taboo subject but usually people do not discuss these topics. To the next! Kind regards!
I have to thank you for the efforts you’ve put in writing this website. I really hope to see the same high-grade content by you in the future as well. In truth, your creative writing abilities has motivated me to get my own, personal blog now 😉
Everything is very open with a precise explanation of the challenges. It was truly informative. Your site is very useful. Thanks for sharing!
Next time I read a blog, I hope that it won’t disappoint me as much as this one. I mean, Yes, it was my choice to read, nonetheless I truly thought you would probably have something useful to say. All I hear is a bunch of crying about something you could possibly fix if you were not too busy searching for attention.
Hi there! I could have sworn I’ve visited your blog before but after going through some of the articles I realized it’s new to me. Anyways, I’m definitely delighted I stumbled upon it and I’ll be book-marking it and checking back regularly!
Aw, this was an exceptionally nice post. Taking the time and actual effort to produce a great article… but what can I say… I procrastinate a lot and don’t manage to get nearly anything done.
Oh my goodness! Impressive article dude! Thanks, However I am having difficulties with your RSS. I don’t understand the reason why I cannot subscribe to it. Is there anybody else having the same RSS problems? Anyone who knows the answer can you kindly respond? Thanks.
When I originally left a comment I appear to have clicked on the -Notify me when new comments are added- checkbox and from now on every time a comment is added I get four emails with the same comment. Is there a way you are able to remove me from that service? Thanks a lot.
I couldn’t refrain from commenting. Very well written.
After looking at a number of the articles on your web page, I seriously appreciate your technique of writing a blog. I bookmarked it to my bookmark site list and will be checking back soon. Take a look at my web site as well and tell me how you feel.
Good day! I just wish to give you a huge thumbs up for your great info you have right here on this post. I’ll be coming back to your web site for more soon.
Greetings! Very useful advice within this post! It is the little changes which will make the most important changes. Many thanks for sharing!
Right here is the right website for anybody who wants to understand this topic. You understand so much its almost tough to argue with you (not that I actually would want to…HaHa). You definitely put a new spin on a topic that’s been discussed for years. Great stuff, just great.
This is the right blog for anyone who hopes to understand this topic. You realize so much its almost tough to argue with you (not that I really would want to…HaHa). You definitely put a brand new spin on a subject that’s been discussed for years. Wonderful stuff, just excellent.
Way cool! Some extremely valid points! I appreciate you penning this article and the rest of the site is really good.
Pretty! This has been a really wonderful article. Thanks for supplying these details.
Hello there! This article could not be written any better! Looking at this post reminds me of my previous roommate! He always kept preaching about this. I most certainly will forward this post to him. Pretty sure he’ll have a very good read. Many thanks for sharing!
Spot on with this write-up, I honestly believe this website needs much more attention. I’ll probably be back again to read through more, thanks for the info!
Very good article! We will be linking to this particularly great article on our website. Keep up the good writing.
Greetings! Very helpful advice within this post! It’s the little changes that produce the most significant changes. Many thanks for sharing!
Excellent article. I definitely love this website. Keep it up!
bookmarked!!, I love your blog.
An impressive share! I’ve just forwarded this onto a friend who has been doing a little homework on this. And he actually ordered me breakfast because I discovered it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanx for spending some time to discuss this subject here on your internet site.
When I originally commented I seem to have clicked the -Notify me when new comments are added- checkbox and from now on every time a comment is added I recieve 4 emails with the same comment. Is there a means you can remove me from that service? Thanks a lot.
There is definately a great deal to learn about this subject. I really like all the points you’ve made.
Very good write-up. I certainly love this website. Keep writing!
Greetings! Very helpful advice within this post! It’s the little changes which will make the most significant changes. Thanks a lot for sharing!
You ought to be a part of a contest for one of the greatest blogs on the web. I most certainly will highly recommend this web site!
Hi, I believe your web site may be having internet browser compatibility issues. Whenever I take a look at your blog in Safari, it looks fine however when opening in I.E., it has some overlapping issues. I just wanted to provide you with a quick heads up! Apart from that, fantastic blog!
Greetings! Very useful advice within this article! It is the little changes that make the biggest changes. Many thanks for sharing!
The next time I read a blog, I hope that it won’t fail me just as much as this particular one. I mean, I know it was my choice to read through, but I really believed you’d have something useful to talk about. All I hear is a bunch of crying about something that you could possibly fix if you weren’t too busy looking for attention.
Aw, this was a very nice post. Taking the time and actual effort to make a good article… but what can I say… I procrastinate a whole lot and never manage to get nearly anything done.
I wanted to thank you for this wonderful read!! I definitely enjoyed every little bit of it. I have got you book marked to look at new things you post…
I’m more than happy to uncover this great site. I want to to thank you for your time just for this wonderful read!! I definitely enjoyed every bit of it and I have you bookmarked to see new things in your blog.
Howdy! This post couldn’t be written much better! Going through this post reminds me of my previous roommate! He continually kept talking about this. I most certainly will forward this post to him. Fairly certain he’ll have a good read. Many thanks for sharing!
This is a topic that is near to my heart… Best wishes! Exactly where can I find the contact details for questions?
This excellent website really has all the information I wanted about this subject and didn’t know who to ask.
Howdy! This blog post could not be written any better! Looking at this post reminds me of my previous roommate! He constantly kept talking about this. I will send this article to him. Pretty sure he’s going to have a very good read. I appreciate you for sharing!
Spot on with this write-up, I truly believe this amazing site needs much more attention. I’ll probably be returning to read more, thanks for the advice!
Hi there! This article could not be written much better! Looking through this article reminds me of my previous roommate! He continually kept preaching about this. I will forward this post to him. Fairly certain he’s going to have a great read. Thanks for sharing!
I’m pretty pleased to find this web site. I want to to thank you for ones time due to this wonderful read!! I definitely liked every part of it and I have you book marked to check out new information on your site.
Great article. I will be facing a few of these issues as well..
An impressive share! I have just forwarded this onto a friend who has been doing a little research on this. And he actually bought me lunch simply because I discovered it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanks for spending some time to discuss this topic here on your internet site.
An impressive share! I have just forwarded this onto a coworker who was conducting a little homework on this. And he in fact bought me dinner because I stumbled upon it for him… lol. So let me reword this…. Thanks for the meal!! But yeah, thanks for spending time to talk about this topic here on your web page.
Good post. I learn something totally new and challenging on blogs I stumbleupon every day. It’s always useful to read through articles from other authors and practice a little something from other sites.
This excellent website definitely has all of the info I needed about this subject and didn’t know who to ask.
This is the right blog for anyone who wishes to understand this topic. You know so much its almost hard to argue with you (not that I really will need to…HaHa). You certainly put a fresh spin on a subject that has been written about for many years. Excellent stuff, just wonderful.
An outstanding share! I’ve just forwarded this onto a coworker who was conducting a little research on this. And he actually bought me dinner because I found it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanks for spending some time to discuss this topic here on your blog.
This is a topic that’s near to my heart… Best wishes! Where can I find the contact details for questions?
I like what you guys are up too. Such clever work and reporting! Carry on the excellent works guys I have incorporated you guys to my blogroll. I think it’ll improve the value of my website 🙂
Howdy! This blog post could not be written any better! Looking through this post reminds me of my previous roommate! He continually kept preaching about this. I will forward this information to him. Fairly certain he’ll have a good read. Many thanks for sharing!
I blog quite often and I genuinely appreciate your content. This great article has truly peaked my interest. I am going to book mark your blog and keep checking for new information about once a week. I opted in for your Feed too.
Das Mirage direkt in der Mitte vom Las Vegas Strip und bietet eine
der bekanntesten Attraktionen von Las Vegas, der Vulkan vor dem Hotel.
Die BARE Pool Lounge für Erwachsene verfügt außerdem über eine Full-Service-Bar und Live-DJs.
Das originale Schild vor dem Hotel ist das größte freistehende Schild der Welt.
Für besonderen Komfort in den Badezimmern sorgen Kosmetikartikel.
Zur sportlichen Betätigung in der Unterbringung stehen den Gästen Yoga und gebührenpflichtig ein Fitnessstudio zur Auswahl.
Ein Whirlpool lockt zur Muskelentspannung und eine Pool-/Snackbar zur Stärkung zwischendurch.
Das Spa bietet eine Vielzahl von Anwendungen sowie Dampfbäder,
Saunen und Whirlpools. Das Mirage verfügt über eine Vielzahl von Funktionen, bei denen Unterhaltung und Spaß im Vordergrund stehen. Die Suiten reichen von der 75,4 Quadratmeter großen Mirage-Suite mit zwei Bädern und einem
Whirlpool über Whirlpool bis zu den 159 Quadratmeter großen Hospitality-Suiten mit einem oder
zwei Schlafzimmern.
Das Badezimmer, ausgestattet mit einer Dusche und einer Badewanne, verfügt über einen Haartrockner.
Die Süßwasserpoolanlage (zum Teil beheizt) bietet erfrischenden Badespaß im
Außenbereich. Ihr Anliegen ist es, Bildung nicht auf eine Vorbereitung für das effiziente Funktionieren im Arbeitsmarkt zu reduzieren, sondern als umfassende
Persönlichkeitsentwicklung und Befähigung zum Engagement
zu verstehen. Eines der ikonischsten Merkmale
des Mirage ist die tägliche Vulkan-Aufführung,
bei der ein künstlicher Vulkan vor dem Resort ausbricht
und ein beeindruckendes Spektakel aus Feuer und Wasser bietet.
„Das Mirage Resort and Casino Las Vegas ist ein Ort, der unglaubliche Erfahrungen bietet und Menschen inspiriert, ihre Träume
zu verfolgen.“ – Steve Wynn, Gründer des Mirage Resorts
References:
https://online-spielhallen.de/spirit-casino-bewertung-ein-umfassender-einblick-fur-spieler/
You ought to take part in a contest for one of the finest blogs on the net. I most certainly will recommend this web site!
Greetings! Very useful advice within this article! It’s the little changes that produce the largest changes. Thanks a lot for sharing!
bookmarked!!, I like your blog!
You are so cool! I do not suppose I have read through a single thing like this before. So good to find somebody with some unique thoughts on this issue. Really.. many thanks for starting this up. This website is one thing that’s needed on the internet, someone with a little originality.
You made some good points there. I checked on the net for more information about the issue and found most people will go along with your views on this website.
I really like looking through an article that will make people think. Also, thanks for allowing me to comment.
11 second baking soda trick for men
Very good article. I absolutely appreciate this website. Keep it up!
Very good article. I will be dealing with many of these issues as well..
That is a good tip particularly to those fresh to the blogosphere. Short but very precise information… Thanks for sharing this one. A must read post!
Aw, this was an extremely nice post. Taking the time and actual effort to produce a top notch article… but what can I say… I hesitate a lot and never seem to get nearly anything done.
I absolutely love your site.. Very nice colors & theme. Did you develop this site yourself? Please reply back as I’m wanting to create my very own website and would love to know where you got this from or what the theme is named. Thanks!
Das Hotel bietet den Gästen auch WLAN in den öffentlichen Bereichen an. Darüber hinaus
ist der Hamburger ZOB nur 350 Meter entfernt,
was für Gäste, die mit dem Bus anreisen, Das Hotel befindet sich in einer sehr
zentralen Lage, nur 350 Meter vom Hamburger Hauptbahnhof entfernt,was die Anreise für Gäste sehr bequem macht.
Du kannst deine Auswahl jederzeit überprüfen und deine Zustimmung zurückziehen, indem du auf
den Link ’Einstellungen zur Privatsphäre’ in der seitlichen Navigation klickst.
Um die einzelnen Verarbeitungszwecke und Cookie-Kategorien zu überprüfen, klicke auf ’Individuelle
Zwecke auswählen’. Du kannst die Anbieter und ihre individuellen Verarbeitungszwecke
in der Anbieterliste überprüfen.
Um wie viel Uhr kann man frühstens im Sleephotels Casino einchecken?
Welcher Service ist im Sleephotels Casino besonders gut
bewertet? Parkmöglichkeiten Die Telefonnummer Sleephotels Casino ist auf unserer Website nicht
verfügbar. Sleephotels Casino Reservierungen unter ‘Zimmer’ möglich
References:
https://online-spielhallen.de/bwin-casino-freispiele-alles-was-sie-wissen-mussen/
I really like reading a post that can make people think. Also, many thanks for permitting me to comment.
I like reading an article that will make men and women think. Also, thank you for allowing for me to comment.
I’m impressed, I must say. Seldom do I encounter a blog that’s equally educative and engaging, and let me tell you, you’ve hit the nail on the head. The problem is an issue that not enough people are speaking intelligently about. Now i’m very happy I stumbled across this during my hunt for something relating to this.
You have made some decent points there. I checked on the internet for more info about the issue and found most individuals will go along with your views on this site.
Howdy! This article could not be written any better! Reading through this post reminds me of my previous roommate! He continually kept preaching about this. I’ll forward this information to him. Pretty sure he will have a very good read. Thanks for sharing!
I was able to find good advice from your blog articles.
I truly love your website.. Very nice colors & theme. Did you make this web site yourself? Please reply back as I’m attempting to create my very own website and would like to find out where you got this from or just what the theme is named. Thank you.
I was very happy to discover this website. I wanted to thank you for ones time just for this wonderful read!! I definitely really liked every bit of it and i also have you book marked to see new information in your blog.
Was die Anzahl betrifft, bietet das Casino zum
Zeitpunkt unseres Immerion Casino Reviews über 500 Titel.
Zudem erhalten Sie einen Überblick darüber, wie
viele Titel von welcher Marke verfügbar sind. Innerhalb der
einzelnen Sparten wurde auf eine Trennung zwischen Live- und RNG Spielen verzichtet,
obwohl erstere zusätzlich einen eigenen Button in der Menüleiste erhalten haben. Die Tischspiele sind auch in der linken Menüleiste unter der gleichnamigen Rubrik
zu finden.
Dieser stand zum Zeitpunkt unseres Besuchs bei knapp 145 USD (bis zu 500 USD sind möglich).
Außerdem gibt es die Möglichkeit, zusätzlich 10 % wöchentliches Cashback zu erhalten. Alternativ
zum klassischen Neukundenbonus gibt es noch ein Highroller Bonus.
Innerhalb dieser Zeit müssen die Freispiele vollständig abgerufen die Umsatzbedingungen erfüllt werden.
Denn neben einem klassischen Willkommensbonus, der mit
8.000€ recht üppig ausfällt und dem VIP-Bonus von bis zu 60.000€, werden alle potenziellen Kunden gleichwohl angesprochen. In unserem
Testbericht gehen wir nun näher auf die Spielauswahl ein, ob es aktuell
gültige Immerion Casino Bonuscodes gibt und an welchen Stellen wir uns noch etwas Nachbesserung wünschen würden. Auch spannende Live Gameshows sind
im Immerion Casino mit am Start, unter anderem in Form
von Extra Chilli Epic Spins, Funky Time, Sweet Bonanza Candyland, Cash or Crash live,
Crazy Time oder Mega Wheel. Man kann also nicht
genau erkennen, wie viele Roulettespiele oder Blackjack Tische vorhanden sind,
da alle Live Spiele in eine einzige große Liste geklatscht wurden. Bei einem erhaltenen Bonusbetrag in Höhe
von 100 Euro würde die Gewinngrenze somit 1.000 Euro
betragen. Zudem gilt es zu beachten, dass mit erhaltenem Bonusguthaben maximal das 10-fache an Gewinnen erspielt werden kann.
References:
https://online-spielhallen.de/ihr-ultimativer-leitfaden-fur-lucky-dreams-casino-bonus-codes/
I like it when individuals come together and share thoughts. Great website, stick with it.
Right here is the perfect site for anybody who would like to understand this topic. You know so much its almost tough to argue with you (not that I personally would want to…HaHa). You certainly put a fresh spin on a topic that’s been discussed for decades. Wonderful stuff, just excellent.
I truly love your website.. Pleasant colors & theme. Did you develop this amazing site yourself? Please reply back as I’m looking to create my own site and would love to find out where you got this from or just what the theme is called. Thanks!
This blog was… how do you say it? Relevant!! Finally I have found something which helped me. Thanks!
May I just say what a comfort to discover an individual who genuinely understands what they’re talking about on the net. You definitely realize how to bring a problem to light and make it important. A lot more people must check this out and understand this side of the story. It’s surprising you are not more popular because you definitely possess the gift.
Howdy! This article could not be written much better! Reading through this post reminds me of my previous roommate! He continually kept preaching about this. I will send this post to him. Pretty sure he will have a great read. Thank you for sharing!
An interesting discussion is worth comment. I do believe that you need to write more about this subject, it may not be a taboo matter but typically people don’t speak about such subjects. To the next! Kind regards!
An outstanding share! I have just forwarded this onto a coworker who was conducting a little research on this. And he in fact ordered me breakfast simply because I discovered it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanks for spending time to talk about this subject here on your website.
I could not resist commenting. Exceptionally well written.
The next time I read a blog, I hope that it does not disappoint me just as much as this one. I mean, I know it was my choice to read through, nonetheless I truly thought you would probably have something interesting to talk about. All I hear is a bunch of whining about something that you could possibly fix if you weren’t too busy looking for attention.
Way cool! Some extremely valid points! I appreciate you writing this article and also the rest of the site is really good.
Great article! We are linking to this great article on our website. Keep up the great writing.
Good article! We are linking to this particularly great post on our website. Keep up the great writing.
When I originally commented I appear to have clicked on the -Notify me when new comments are added- checkbox and from now on whenever a comment is added I recieve 4 emails with the exact same comment. There has to be an easy method you are able to remove me from that service? Thanks.
This is a good tip especially to those new to the blogosphere. Simple but very accurate info… Thanks for sharing this one. A must read article!
I’m pretty pleased to find this web site. I wanted to thank you for ones time due to this wonderful read!! I definitely liked every little bit of it and i also have you saved as a favorite to see new things on your web site.
Your style is very unique compared to other people I’ve read stuff from. I appreciate you for posting when you’ve got the opportunity, Guess I’ll just bookmark this page.
An interesting discussion is worth comment. I do believe that you need to publish more about this subject, it might not be a taboo subject but typically people don’t discuss these subjects. To the next! Cheers.
Hi there! I could have sworn I’ve been to this website before but after looking at many of the articles I realized it’s new to me. Regardless, I’m definitely pleased I stumbled upon it and I’ll be bookmarking it and checking back regularly!
This excellent website certainly has all the information and facts I wanted concerning this subject and didn’t know who to ask.
Hello there! I could have sworn I’ve visited this website before but after going through many of the articles I realized it’s new to me. Regardless, I’m definitely pleased I stumbled upon it and I’ll be bookmarking it and checking back frequently.
I enjoy reading through a post that can make men and women think. Also, many thanks for allowing for me to comment.
You made some good points there. I checked on the net for additional information about the issue and found most individuals will go along with your views on this website.
I’m extremely pleased to discover this site. I want to to thank you for your time for this wonderful read!! I definitely enjoyed every part of it and I have you book-marked to look at new information on your website.
May I simply say what a relief to uncover an individual who actually knows what they are discussing over the internet. You certainly realize how to bring an issue to light and make it important. More and more people ought to look at this and understand this side of the story. It’s surprising you aren’t more popular given that you definitely possess the gift.
Pretty! This has been an extremely wonderful post. Thank you for supplying these details.
Very good post. I will be dealing with a few of these issues as well..
Good web site you’ve got here.. It’s difficult to find quality writing like yours nowadays. I really appreciate individuals like you! Take care!!
I love it when folks come together and share ideas. Great website, stick with it.
Hi, I do believe this is an excellent site. I stumbledupon it 😉 I may revisit once again since i have saved as a favorite it. Money and freedom is the greatest way to change, may you be rich and continue to guide others.
Hi there! This blog post couldn’t be written much better! Looking through this post reminds me of my previous roommate! He constantly kept talking about this. I am going to send this information to him. Fairly certain he’ll have a very good read. Thanks for sharing!
I want to to thank you for this great read!! I certainly enjoyed every bit of it. I have you book marked to check out new things you post…
Wonderful article! We will be linking to this great content on our website. Keep up the great writing.
Hi there! This article could not be written any better! Reading through this article reminds me of my previous roommate! He continually kept preaching about this. I most certainly will send this post to him. Pretty sure he’s going to have a good read. Thank you for sharing!
Greetings! Very helpful advice in this particular post! It’s the little changes that produce the most significant changes. Many thanks for sharing!
This is a topic that is near to my heart… Thank you! Exactly where can I find the contact details for questions?
I needed to thank you for this good read!! I absolutely enjoyed every bit of it. I have got you book marked to look at new stuff you post…
I want to to thank you for this wonderful read!! I absolutely enjoyed every little bit of it. I’ve got you book-marked to look at new stuff you post…
Say, you got a nice post.Really thank you! Great.
Aw, this was an exceptionally good post. Taking the time and actual effort to make a really good article… but what can I say… I hesitate a lot and don’t seem to get anything done.
You ought to be a part of a contest for one of the finest blogs on the net. I most certainly will highly recommend this site!
Pretty! This was a really wonderful article. Thank you for providing this information.
Hello there! This article could not be written much better! Looking at this post reminds me of my previous roommate! He continually kept talking about this. I’ll send this information to him. Pretty sure he’ll have a very good read. Thank you for sharing!
Pretty! This has been an incredibly wonderful post. Thanks for providing this info.
Great article. I am going through some of these issues as well..
Spot on with this write-up, I truly believe this amazing site needs far more attention. I’ll probably be back again to read through more, thanks for the info!
This excellent website really has all of the info I wanted concerning this subject and didn’t know who to ask.
Everything is very open with a really clear explanation of the challenges. It was definitely informative. Your website is very useful. Thank you for sharing.
After looking into a number of the blog articles on your web site, I really like your technique of blogging. I added it to my bookmark site list and will be checking back in the near future. Please visit my website as well and let me know what you think.
Good post. I learn something new and challenging on websites I stumbleupon everyday. It will always be exciting to read through content from other writers and practice a little something from their web sites.
Pretty! This was an extremely wonderful post. Thank you for supplying these details.
After exploring a number of the articles on your web site, I truly like your technique of writing a blog. I book marked it to my bookmark site list and will be checking back soon. Please visit my website as well and let me know how you feel.
Saved as a favorite, I really like your web site!
Nice post. I learn something new and challenging on websites I stumbleupon every day. It’s always helpful to read through articles from other authors and practice something from their web sites.
Everyone loves it when people come together and share views. Great site, continue the good work!
It’s nearly impossible to find well-informed people in this particular topic, however, you seem like you know what you’re talking about! Thanks
This is the perfect web site for everyone who really wants to understand this topic. You know so much its almost tough to argue with you (not that I actually would want to…HaHa). You certainly put a new spin on a topic which has been written about for decades. Great stuff, just excellent.
Good post. I learn something totally new and challenging on blogs I stumbleupon on a daily basis. It will always be exciting to read through content from other writers and practice a little something from other sites.
Way cool! Some very valid points! I appreciate you writing this article plus the rest of the site is extremely good.
After going over a few of the blog articles on your blog, I truly like your way of writing a blog. I book-marked it to my bookmark webpage list and will be checking back soon. Please visit my website as well and let me know what you think.
Hi! I simply wish to offer you a big thumbs up for the great info you’ve got right here on this post. I am coming back to your site for more soon.
After exploring a number of the blog articles on your web page, I honestly like your technique of blogging. I bookmarked it to my bookmark website list and will be checking back in the near future. Please visit my website too and let me know what you think.
Greetings! Very useful advice within this post! It’s the little changes that make the biggest changes. Thanks for sharing!
Pretty! This has been a really wonderful article. Thank you for supplying this info.
I like reading a post that will make people think. Also, thanks for allowing me to comment.
This blog was… how do you say it? Relevant!! Finally I’ve found something which helped me. Kudos.
I really love your blog.. Excellent colors & theme. Did you build this web site yourself? Please reply back as I’m attempting to create my very own website and would like to know where you got this from or just what the theme is called. Kudos!
Can I simply just say what a comfort to find someone who genuinely knows what they’re talking about on the internet. You actually realize how to bring a problem to light and make it important. More and more people really need to check this out and understand this side of the story. I can’t believe you’re not more popular because you surely possess the gift.
Many platforms offer a range of progressive jackpot slots,
each with its unique theme and prize amount. Unlike regular slots, where the jackpot is fixed, progressive
jackpots increase with every bet on the game. The presence of secure payment methods further enhances the overall experience.
To recap, choosing the right site to create a casino account at
involves evaluating specific factors. Please note that you can only play in the state
indicated in the first column. The legality of internet gambling in the US is determined on a state-by-state basis.
It typically takes about 2-3 business days for withdrawal requests to process
and arrive in your verified online wallet, bank account,
or Play+ card. You’ll need a valid, government-issued photo ID
when using the in-person method for transactions to and from your online account.
BetMGM Casino has a 24/7 live chat module,
a detailed FAQ section, and a secure email address that’s used for submitting KYC/AML documents along
with Source of Wealth information. There’s really nothing NOT to like
about the platform, as it checks all the important boxes.
You will soon be redirected to the casino’s website. If you can’t stop gambling when you want
to, it’s time to get help.
References:
https://blackcoin.co/best-australian-online-casinos-aussie-gambling-sites-in-2025/
This is the right blog for anyone who really wants to understand this topic. You realize a whole lot its almost hard to argue with you (not that I personally will need to…HaHa). You definitely put a fresh spin on a subject that has been discussed for ages. Excellent stuff, just wonderful.
Winds south to southwesterly 15 to 20 km/h tending south to southeasterly 20 to 30 km/h during the morning then becoming light during the evening.
Overnight temperatures falling to between 13 and
16 with daytime temperatures reaching the mid to
high 20s. Overnight temperatures falling to around 16 with
daytime temperatures reaching the low to mid 20s. Winds southeasterly 20 to 30 km/h turning southerly
15 to 25 km/h in the evening. Winds S 15 to 25 km/h tending SE in the middle of
the day then becoming light in the late evening. Unlock more weather
data and layers options.
It is located 726 km (451 mi) north of Sydney and 228 km (142 mi) south
of Brisbane.
More detailed data for individual sites can be obtained by contacting the Bureau.
Monthly statistics are only included if there are more
than 10 years of data. More detailed data for individual sites is available.
Medium chance of showers in the north, slight chance elsewhere.
References:
https://blackcoin.co/vegastars-casino-your-ultimate-gaming-destination/
That is a good tip particularly to those new to the blogosphere. Brief but very precise information… Appreciate your sharing this one. A must read post.
The table below sums up the essentials so you can quickly decide whether 21Bit fits your playing style and
risk tolerance. We also offer you generous
bonus offers at every turn in your gambling journey. Once you’ve selected your preferred cryptocurrency, you can copy your private depositing address and paste it into your crypto wallet.
21bit strives to become the best crypto casino of all times, running great offers and a fantastic cast of games so that no player
can resist the lure. New players receive a casino sign up bonus of AU$1,000 + 250 Free Spins, split across their first five deposits.
Join 21bit Casino, a secure online casino offering the best slots, immersive live dealer games, and promotions that reward you.
Under a Curaçao licence, offering Australian players a large
selection of slots, live tables and instant payments in digital currencies.
21bit is a go-to crypto casino site that excels at offering generous bonuses and top-notch
crypto gambling experiences. Our game collections range from slots, live casinos and jackpot games to table games.
Additionally, you can get the second, third and even fourth deposit bonuses when you make subsequent deposits
into your account. In short, 21bit Casino bonuses are for every
player’s taste.
References:
https://blackcoin.co/aussie-casinos-australias-largest-online-casino-database/
Highly recommend for anyone looking for a fun, legit sweepstakes casino.
The slots run perfectly on my iPhone, and the site is super fast.
I redeemed my Sweeps Coins for cash and had the money in my
bank account within 48 hours! I signed up, got my free coins, and hit a jackpot within days.
Play using Gold Coins for fun or Sweeps Coins for the chance to redeem real cash prizes,
100% legally in most U.S. states.Get started with 100,000 Gold Coins + 2 FREE Sweeps Coins when you sign up.
The platform employs state-of-the-art encryption technologies to ensure that all
user data is protected against unauthorized access. The
verification process at Crown Casino is a crucial step to ensure the security and integrity
of the platform. To log in to your Crown Casino account, start
by opening the Crown Casino app on your mobile
device or visiting the Crown Casino website using your browser.
The platform prioritizes user convenience, providing clear instructions and an intuitive interface that
makes it easy to get started. You must click on the No Bonus button if
you do not want to activate the welcome bonus.
Instead, there is a welcome bonus button at the top of the deposit screen.
References:
https://blackcoin.co/no-deposit-casino-bonuses-for-australia-free-spins-signup-cash-offers/
Aw, this was a really nice post. Taking the time and actual effort to create a really good article… but what can I say… I put things off a whole lot and never seem to get anything done.
I wanted to thank you for this good read!! I definitely loved every bit of it. I have you bookmarked to look at new things you post…
Hello there, I do think your site could possibly be having browser compatibility problems. Whenever I take a look at your site in Safari, it looks fine however, when opening in I.E., it’s got some overlapping issues. I merely wanted to provide you with a quick heads up! Apart from that, fantastic site.
As you can tell, online casinos go the extra mile these days to satisfy the
needs of their mobile users. Ever since our smartphones have
evolved to run high-end graphics, online casinos have jumped at the opportunity to provide their services on mobile devices.
Online gambling is purely a recreational feat, whether you’re playing for free or for real money.
All of the laws and restrictions regarding online gambling are
aimed at the operators, not the players. Playing on your phone is a basic feature that top real money casinos
should offer.
Fifteen real money online casinos have launched since Michigan lawmakers legalized online casinos, online poker, and online sports betting in 2019.
The Pai Gow Poker variant featuring the Fortune side
bet is featured on many real money online casinos.
While most online casinos provide baccarat, they usually offer fewer variations compared to other table games.
We consider a wide range of factors when creating our list of the
best real money online casinos. If real money online casinos aren’t legal in your state,
be sure to check out the best social casinos and sweepstakes casinos available throughout the United States.
The real money online casinos we recommend are legal and licensed with oversight from state regulatory
agencies.
References:
https://blackcoin.co/45_exclusive-new-jersey-online-poker-vip-programs-compared_rewrite_1/
Hi, I do think this is a great blog. I stumbledupon it 😉 I will return yet again since I book marked it. Money and freedom is the best way to change, may you be rich and continue to help other people.
Good day! I could have sworn I’ve visited this web site before but after going through many of the articles I realized it’s new to me. Anyways, I’m certainly happy I came across it and I’ll be book-marking it and checking back regularly!
Having read this I believed it was very enlightening. I appreciate you taking the time and effort to put this short article together. I once again find myself spending a significant amount of time both reading and leaving comments. But so what, it was still worthwhile!
There is definately a lot to know about this issue. I love all the points you made.
Your style is very unique in comparison to other people I have read stuff from. I appreciate you for posting when you have the opportunity, Guess I’ll just book mark this site.
An interesting discussion is worth comment. I do think that you should write more on this subject matter, it may not be a taboo subject but typically folks don’t speak about such issues. To the next! Kind regards.
Following criticisms of its implementation of those systems, YouTube started treating all videos designated as “made for kids” as liable under COPPA on January 6, 2020.
By February 2017, one billion hours of YouTube videos were being
watched every day, and 400 hours worth of videos were uploaded
every minute. In April 2010, Lady Gaga’s “Bad Romance” became the most-viewed video, becoming the first video to reach
200 million views on May 9, 2010. Hurley announced that he would
be stepping down as chief executive officer of YouTube
to take an advisory role and that Salar Kamangar would take over as head of
the company in October 2010.
While some users praised the move as a way to discourage trolls, others felt that
hiding dislikes would make it harder for viewers to recognize clickbait or unhelpful videos and that other features already existed for creators to limit bullying.
It features a simplified user interface, curated selections
of channels featuring age-appropriate content, and parental control features.
YouTube also released YouTube Music, a third app oriented towards streaming
and discovering the music content hosted on the
YouTube platform.
References:
https://blackcoin.co/stake-casino-australia-where-online-thrills-begin/
Oh my goodness! Incredible article dude! Thank you so much, However I am going through issues with your RSS. I don’t know why I can’t join it. Is there anyone else getting similar RSS issues? Anybody who knows the answer can you kindly respond? Thanx!
You are so cool! I do not suppose I’ve truly read a single thing like that before. So nice to find somebody with some unique thoughts on this issue. Really.. thank you for starting this up. This site is one thing that is required on the web, someone with some originality.
Good web site you’ve got here.. It’s difficult to find quality writing like yours nowadays. I seriously appreciate individuals like you! Take care!!
Great post. I’m facing a few of these issues as well..
This is a topic which is close to my heart… Cheers! Exactly where can I find the contact details for questions?
You’re so interesting! I do not suppose I have read through anything like this before. So good to discover another person with original thoughts on this subject matter. Seriously.. many thanks for starting this up. This web site is one thing that is needed on the internet, someone with some originality.
Howdy! I simply want to offer you a big thumbs up for your great information you’ve got right here on this post. I am returning to your web site for more soon.
I’m amazed, I have to admit. Rarely do I encounter a blog that’s both equally educative and interesting, and without a doubt, you have hit the nail on the head. The issue is something that not enough folks are speaking intelligently about. Now i’m very happy I came across this in my hunt for something concerning this.
Very good info. Lucky me I discovered your site by chance (stumbleupon). I’ve saved it for later.
Hi there! I could have sworn I’ve visited your blog before but after going through a few of the posts I realized it’s new to me. Anyhow, I’m certainly pleased I discovered it and I’ll be book-marking it and checking back often.
Pretty! This has been an incredibly wonderful post. Many thanks for providing these details.
Very good write-up. I absolutely love this website. Thanks!
I blog often and I really appreciate your content. This article has really peaked my interest. I am going to take a note of your site and keep checking for new details about once per week. I opted in for your RSS feed too.
I would like to thank you for the efforts you have put in penning this blog. I am hoping to check out the same high-grade blog posts by you later on as well. In fact, your creative writing abilities has inspired me to get my own, personal website now 😉
Good post. I learn something new and challenging on sites I stumbleupon everyday. It will always be useful to read content from other authors and practice a little something from other websites.
After exploring a handful of the articles on your web page, I seriously appreciate your way of blogging. I added it to my bookmark webpage list and will be checking back in the near future. Take a look at my web site too and tell me what you think.
Excellent site you have got here.. It’s hard to find high quality writing like yours these days. I seriously appreciate people like you! Take care!!
An impressive share! I’ve just forwarded this onto a colleague who has been conducting a little homework on this. And he actually ordered me dinner due to the fact that I discovered it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanks for spending time to discuss this matter here on your blog.
Very good information. Lucky me I recently found your blog by chance (stumbleupon). I’ve saved it for later.
NERVITAL REVIEW
bookmarked!!, I like your blog.
You are so cool! I don’t believe I’ve read through something like this before. So great to find another person with original thoughts on this issue. Really.. many thanks for starting this up. This site is one thing that’s needed on the web, someone with some originality.
I was able to find good advice from your blog articles.
This site was… how do I say it? Relevant!! Finally I have found something that helped me. Thanks.
Spot on with this write-up, I absolutely think this site needs far more attention. I’ll probably be returning to read more, thanks for the info!
You have made some really good points there. I checked on the web to learn more about the issue and found most individuals will go along with your views on this web site.
Hi there! I simply want to give you a big thumbs up for the great info you have right here on this post. I will be returning to your site for more soon.
You’ve made some decent points there. I looked on the internet to find out more about the issue and found most individuals will go along with your views on this site.
This is a topic which is close to my heart… Thank you! Where can I find the contact details for questions?
Aw, this was a really good post. Taking the time and actual effort to produce a superb article… but what can I say… I hesitate a whole lot and never seem to get anything done.
The next time I read a blog, I hope that it won’t disappoint me just as much as this particular one. After all, Yes, it was my choice to read through, but I genuinely thought you’d have something interesting to talk about. All I hear is a bunch of moaning about something that you can fix if you weren’t too busy searching for attention.
I want to to thank you for this excellent read!! I absolutely loved every bit of it. I have got you bookmarked to check out new things you post…
Hi there! This article could not be written any better! Looking through this article reminds me of my previous roommate! He continually kept talking about this. I’ll forward this article to him. Pretty sure he’s going to have a great read. I appreciate you for sharing!
After checking out a handful of the articles on your web site, I honestly appreciate your technique of blogging. I saved it to my bookmark website list and will be checking back in the near future. Please visit my web site as well and let me know how you feel.
Hi there, I believe your web site might be having web browser compatibility problems. Whenever I look at your web site in Safari, it looks fine however, if opening in I.E., it has some overlapping issues. I simply wanted to give you a quick heads up! Apart from that, wonderful website!
It’s nearly impossible to find knowledgeable people for this subject, however, you sound like you know what you’re talking about! Thanks
Aw, this was an exceptionally nice post. Taking the time and actual effort to make a good article… but what can I say… I hesitate a lot and never seem to get nearly anything done.
Hi, I do believe this is an excellent website. I stumbledupon it 😉 I’m going to revisit yet again since I bookmarked it. Money and freedom is the greatest way to change, may you be rich and continue to guide others.
I must thank you for the efforts you’ve put in writing this site. I am hoping to view the same high-grade content by you in the future as well. In truth, your creative writing abilities has encouraged me to get my own, personal site now 😉
I’d like to thank you for the efforts you have put in penning this blog. I’m hoping to see the same high-grade blog posts from you in the future as well. In truth, your creative writing abilities has inspired me to get my own, personal website now 😉
I have to thank you for the efforts you have put in writing this blog. I really hope to see the same high-grade content from you later on as well. In truth, your creative writing abilities has motivated me to get my very own website now 😉
paypal casino usa
References:
ww.enhasusg.co.kr
paypal casino uk
References:
https://workfind.in/profile/eldenbejah5097
online casino accepts paypal us
References:
https://vhembedirect.co.za/employer/best-online-casinos-in-australia-top-casino-sites-for-2025/
online poker real money paypal
References:
https://working.altervista.org/employer/best-paypal-casinos-updated-2025/
Pretty! This was an extremely wonderful post. Thank you for supplying this info.
Howdy very cool website!! Man .. Beautiful .. Superb .. I’ll bookmark your site and take the feeds additionally…I’m satisfied to find numerous helpful info here in the publish, we need work out extra techniques in this regard, thanks for sharing.
paypal casino usa
References:
https://jobhaiti.net/employer/20-best-online-casinos-in-australia-for-real-money-in-2025/
online casino real money paypal
References:
http://www.grapvocar.site/bbs/board.php?bo_table=free&wr_id=1788
online betting with paypal winnersbet
References:
https://cvbankye.com/employer/best-online-casinos-australia-2025-top-aussie-casino-sites/
online slot machines paypal
References:
https://walsallads.co.uk/profile/karmakingsmill
That is a great tip especially to those fresh to the blogosphere. Brief but very accurate info… Many thanks for sharing this one. A must read article!
Hi, I do believe this is a great web site. I stumbledupon it 😉 I am going to revisit yet again since I book marked it. Money and freedom is the best way to change, may you be rich and continue to help others.
There is definately a great deal to know about this issue. I love all of the points you made.
I quite like reading a post that can make men and women think. Also, thanks for allowing me to comment.
Having read this I thought it was really informative. I appreciate you spending some time and energy to put this article together. I once again find myself spending way too much time both reading and leaving comments. But so what, it was still worth it.
Pretty! This was a really wonderful article. Thank you for supplying this information.
This site was… how do you say it? Relevant!! Finally I have found something that helped me. Appreciate it!
This is the right blog for anyone who really wants to find out about this topic. You realize so much its almost tough to argue with you (not that I really would want to…HaHa). You definitely put a brand new spin on a subject which has been discussed for ages. Wonderful stuff, just wonderful.
This is a great tip particularly to those fresh to the blogosphere. Short but very accurate information… Thank you for sharing this one. A must read article!
I couldn’t resist commenting. Well written.
This blog was… how do I say it? Relevant!! Finally I have found something that helped me. Thank you.
An impressive share! I’ve just forwarded this onto a coworker who was doing a little research on this. And he actually ordered me dinner due to the fact that I discovered it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanx for spending some time to talk about this topic here on your web page.
Aw, this was a really nice post. Taking the time and actual effort to produce a good article… but what can I say… I put things off a whole lot and don’t manage to get anything done.
Everything is very open with a really clear clarification of the challenges. It was really informative. Your website is useful. Thank you for sharing.
I need to to thank you for this fantastic read!! I definitely loved every bit of it. I’ve got you book marked to check out new things you post…
This is a topic which is near to my heart… Cheers! Exactly where are your contact details though?
It’s hard to find knowledgeable people about this topic, but you sound like you know what you’re talking about! Thanks
Excellent article. I will be facing many of these issues as well..
This web site really has all the info I wanted about this subject and didn’t know who to ask.
I seriously love your blog.. Great colors & theme. Did you develop this website yourself? Please reply back as I’m wanting to create my very own website and want to know where you got this from or what the theme is called. Thanks!
Everyone loves it when people come together and share opinions. Great site, continue the good work.
Aw, this was an incredibly good post. Spending some time and actual effort to generate a really good article… but what can I say… I hesitate a lot and never seem to get anything done.
Spot on with this write-up, I seriously think this website needs far more attention. I’ll probably be back again to see more, thanks for the information.
I’m amazed, I must say. Rarely do I encounter a blog that’s both educative and interesting, and let me tell you, you’ve hit the nail on the head. The issue is something which too few people are speaking intelligently about. I’m very happy that I stumbled across this during my hunt for something relating to this.
Having read this I believed it was really enlightening. I appreciate you spending some time and effort to put this short article together. I once again find myself personally spending a lot of time both reading and posting comments. But so what, it was still worth it.
This is the right web site for everyone who would like to find out about this topic. You know a whole lot its almost hard to argue with you (not that I really would want to…HaHa). You certainly put a new spin on a subject that has been written about for decades. Wonderful stuff, just wonderful.
Hi, I do believe this is an excellent website. I stumbledupon it 😉 I’m going to return yet again since i have saved as a favorite it. Money and freedom is the greatest way to change, may you be rich and continue to help other people.
An outstanding share! I have just forwarded this onto a coworker who was conducting a little homework on this. And he actually ordered me lunch simply because I discovered it for him… lol. So allow me to reword this…. Thank YOU for the meal!! But yeah, thanks for spending some time to discuss this issue here on your site.
I seriously love your website.. Great colors & theme. Did you make this amazing site yourself? Please reply back as I’m attempting to create my own blog and would like to learn where you got this from or exactly what the theme is named. Many thanks.
You’ve made some good points there. I checked on the internet for more information about the issue and found most people will go along with your views on this site.
Nice post. I learn something new and challenging on sites I stumbleupon everyday. It’s always useful to read through content from other authors and use a little something from their web sites.
This is a topic which is near to my heart… Cheers! Exactly where can I find the contact details for questions?
Excellent blog you have here.. It’s difficult to find high quality writing like yours nowadays. I seriously appreciate individuals like you! Take care!!
I really love your blog.. Very nice colors & theme. Did you make this amazing site yourself? Please reply back as I’m looking to create my own website and would love to find out where you got this from or just what the theme is named. Thank you!
An intriguing discussion is worth comment. I think that you ought to publish more on this issue, it might not be a taboo subject but typically folks don’t talk about such subjects. To the next! Best wishes!
Spot on with this write-up, I really feel this website needs a lot more attention. I’ll probably be back again to read through more, thanks for the information.
I need to to thank you for this very good read!! I absolutely loved every bit of it. I have got you saved as a favorite to look at new things you post…
I really like reading an article that can make people think. Also, many thanks for permitting me to comment.
bookmarked!!, I like your site.
Hi! I simply would like to offer you a big thumbs up for the great info you have here on this post. I’ll be returning to your web site for more soon.
I blog often and I really appreciate your content. The article has really peaked my interest. I’m going to book mark your site and keep checking for new details about once a week. I opted in for your RSS feed too.
This is the perfect site for everyone who hopes to find out about this topic. You know a whole lot its almost hard to argue with you (not that I personally would want to…HaHa). You definitely put a fresh spin on a topic which has been written about for a long time. Excellent stuff, just wonderful.
That is a really good tip particularly to those new to the blogosphere. Brief but very accurate information… Thanks for sharing this one. A must read article.
This is a really good tip particularly to those fresh to the blogosphere. Short but very precise info… Appreciate your sharing this one. A must read post.
Your style is so unique in comparison to other folks I have read stuff from. I appreciate you for posting when you’ve got the opportunity, Guess I’ll just book mark this site.
I quite like looking through an article that can make men and women think. Also, many thanks for permitting me to comment.
Howdy! I just would like to offer you a huge thumbs up for the great info you’ve got right here on this post. I am coming back to your blog for more soon.
I’m more than happy to find this page. I want to to thank you for ones time due to this wonderful read!! I definitely liked every part of it and i also have you book marked to check out new stuff in your blog.
I must thank you for the efforts you have put in writing this website. I’m hoping to check out the same high-grade content from you in the future as well. In fact, your creative writing abilities has motivated me to get my own, personal site now 😉
Very nice write-up. I definitely love this site. Continue the good work!
Greetings! Very helpful advice in this particular post! It is the little changes that make the largest changes. Thanks a lot for sharing!
Great article! We are linking to this great content on our site. Keep up the good writing.
This is a topic that is near to my heart… Many thanks! Exactly where can I find the contact details for questions?
Right here is the right webpage for anyone who wishes to understand this topic. You understand a whole lot its almost tough to argue with you (not that I personally would want to…HaHa). You certainly put a fresh spin on a subject that has been discussed for ages. Great stuff, just great.
There is definately a great deal to know about this topic. I love all of the points you have made.
May I simply just say what a comfort to discover a person that actually knows what they’re talking about over the internet. You actually realize how to bring an issue to light and make it important. More and more people have to read this and understand this side of your story. It’s surprising you aren’t more popular given that you surely possess the gift.
You need to be a part of a contest for one of the highest quality blogs on the web. I most certainly will highly recommend this blog!
This is a great tip especially to those new to the blogosphere. Brief but very accurate information… Thank you for sharing this one. A must read article!
When I originally left a comment I appear to have clicked the -Notify me when new comments are added- checkbox and now whenever a comment is added I get 4 emails with the exact same comment. There has to be a way you are able to remove me from that service? Many thanks.
After going over a number of the articles on your site, I seriously appreciate your way of writing a blog. I book-marked it to my bookmark website list and will be checking back in the near future. Please visit my web site as well and let me know how you feel.
There is definately a great deal to know about this issue. I like all the points you made.
Great information. Lucky me I discovered your site by chance (stumbleupon). I’ve saved as a favorite for later.
It’s nearly impossible to find knowledgeable people in this particular topic, however, you sound like you know what you’re talking about! Thanks
Greetings! Very useful advice within this article! It’s the little changes that make the largest changes. Thanks for sharing!
An outstanding share! I have just forwarded this onto a co-worker who has been conducting a little research on this. And he in fact ordered me breakfast due to the fact that I found it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanks for spending the time to talk about this matter here on your site.
Excellent article! We are linking to this particularly great post on our site. Keep up the good writing.
This site was… how do I say it? Relevant!! Finally I’ve found something which helped me. Many thanks.
It’s difficult to find knowledgeable people for this subject, but you sound like you know what you’re talking about! Thanks
Spot on with this write-up, I really believe that this website needs a lot more attention. I’ll probably be returning to read more, thanks for the info!
Everything is very open with a really clear description of the issues. It was truly informative. Your website is very useful. Many thanks for sharing!
Hello! I just want to offer you a big thumbs up for the excellent info you’ve got right here on this post. I’ll be returning to your blog for more soon.
I like it when people get together and share thoughts. Great site, stick with it.
Wonderful article! We are linking to this great post on our website. Keep up the good writing.
After looking into a number of the blog articles on your web page, I honestly appreciate your technique of writing a blog. I book marked it to my bookmark webpage list and will be checking back in the near future. Please check out my website as well and tell me what you think.
Excellent post. I definitely love this website. Keep it up!
Hello there, I think your blog may be having internet browser compatibility issues. When I look at your web site in Safari, it looks fine however, when opening in Internet Explorer, it has some overlapping issues. I merely wanted to give you a quick heads up! Aside from that, wonderful blog.
You’ve made some really good points there. I looked on the net to find out more about the issue and found most people will go along with your views on this website.
Can I simply say what a relief to discover someone who genuinely understands what they are talking about on the net. You definitely realize how to bring a problem to light and make it important. More and more people ought to check this out and understand this side of the story. I can’t believe you aren’t more popular given that you certainly have the gift.
Good info. Lucky me I ran across your website by chance (stumbleupon). I have bookmarked it for later.
You should be a part of a contest for one of the best sites on the internet. I’m going to highly recommend this site!
Very good article. I certainly love this website. Keep writing!
Spot on with this write-up, I truly feel this amazing site needs much more attention. I’ll probably be returning to read through more, thanks for the advice.
I couldn’t resist commenting. Exceptionally well written!
After I originally left a comment I appear to have clicked the -Notify me when new comments are added- checkbox and now every time a comment is added I recieve four emails with the same comment. Is there a way you are able to remove me from that service? Thanks.
Way cool! Some extremely valid points! I appreciate you penning this article and the rest of the website is also really good.
It’s difficult to find educated people in this particular topic, but you sound like you know what you’re talking about! Thanks
Hi! I could have sworn I’ve visited your blog before but after browsing through a few of the posts I realized it’s new to me. Nonetheless, I’m certainly happy I came across it and I’ll be bookmarking it and checking back often.
Spot on with this write-up, I honestly believe this web site needs a great deal more attention. I’ll probably be returning to see more, thanks for the advice!
When I originally left a comment I seem to have clicked the -Notify me when new comments are added- checkbox and now every time a comment is added I get four emails with the exact same comment. Perhaps there is an easy method you can remove me from that service? Cheers.
Howdy! I could have sworn I’ve been to your blog before but after going through a few of the articles I realized it’s new to me. Nonetheless, I’m definitely happy I stumbled upon it and I’ll be bookmarking it and checking back frequently!
Having read this I believed it was very enlightening. I appreciate you spending some time and effort to put this informative article together. I once again find myself personally spending way too much time both reading and commenting. But so what, it was still worthwhile.
Great site you’ve got here.. It’s hard to find high quality writing like yours nowadays. I honestly appreciate individuals like you! Take care!!
I was able to find good information from your blog articles.
Great web site you’ve got here.. It’s difficult to find good quality writing like yours these days. I truly appreciate people like you! Take care!!
I could not refrain from commenting. Very well written.
I love it whenever people come together and share thoughts. Great blog, keep it up.
Greetings, I believe your blog could possibly be having web browser compatibility problems. When I look at your web site in Safari, it looks fine but when opening in IE, it has some overlapping issues. I merely wanted to provide you with a quick heads up! Aside from that, excellent blog!
There’s certainly a great deal to know about this subject. I love all of the points you’ve made.
Oh my goodness! Impressive article dude! Many thanks, However I am encountering problems with your RSS. I don’t know why I cannot subscribe to it. Is there anybody getting identical RSS issues? Anyone that knows the solution will you kindly respond? Thanx!
This site really has all of the info I wanted concerning this subject and didn’t know who to ask.
This is the right web site for anybody who wishes to find out about this topic. You realize a whole lot its almost hard to argue with you (not that I really will need to…HaHa). You certainly put a brand new spin on a subject which has been written about for ages. Wonderful stuff, just wonderful.
Having read this I thought it was really enlightening. I appreciate you spending some time and energy to put this informative article together. I once again find myself spending way too much time both reading and posting comments. But so what, it was still worthwhile.
You ought to take part in a contest for one of the greatest blogs on the web. I’m going to recommend this web site!
You are so interesting! I don’t suppose I’ve truly read a single thing like this before. So wonderful to find another person with some genuine thoughts on this topic. Seriously.. thank you for starting this up. This site is something that’s needed on the internet, someone with a little originality.
You are so interesting! I do not suppose I’ve read through a single thing like this before. So wonderful to discover someone with original thoughts on this issue. Seriously.. many thanks for starting this up. This web site is something that is needed on the internet, someone with some originality.
I’m impressed, I must say. Seldom do I come across a blog that’s both equally educative and engaging, and let me tell you, you have hit the nail on the head. The problem is something that not enough people are speaking intelligently about. I’m very happy I found this during my hunt for something regarding this.
Hello, There’s no doubt that your web site could be having browser compatibility issues. When I look at your blog in Safari, it looks fine however, when opening in IE, it’s got some overlapping issues. I just wanted to provide you with a quick heads up! Aside from that, excellent website.
Wonderful article! We are linking to this particularly great post on our website. Keep up the great writing.
Hi! I could have sworn I’ve been to this web site before but after looking at a few of the posts I realized it’s new to me. Anyways, I’m certainly delighted I stumbled upon it and I’ll be book-marking it and checking back frequently.
You have made some good points there. I looked on the net for additional information about the issue and found most individuals will go along with your views on this website.
This page truly has all the info I wanted concerning this subject and didn’t know who to ask.
Right here is the perfect website for anyone who hopes to understand this topic. You know a whole lot its almost tough to argue with you (not that I personally would want to…HaHa). You definitely put a fresh spin on a subject that’s been written about for years. Great stuff, just excellent.
Howdy! This article couldn’t be written much better! Reading through this post reminds me of my previous roommate! He continually kept talking about this. I’ll forward this article to him. Fairly certain he’s going to have a great read. Thank you for sharing!
I have to thank you for the efforts you have put in penning this blog. I’m hoping to see the same high-grade blog posts from you later on as well. In fact, your creative writing abilities has motivated me to get my own site now 😉
I love reading an article that will make people think. Also, thank you for allowing me to comment.
This is a topic that’s near to my heart… Cheers! Exactly where can I find the contact details for questions?
I like it when folks come together and share opinions. Great blog, continue the good work!
Everything is very open with a clear description of the issues. It was really informative. Your website is very useful. Thanks for sharing.
Hello there, There’s no doubt that your site may be having browser compatibility problems. When I look at your website in Safari, it looks fine however when opening in Internet Explorer, it’s got some overlapping issues. I just wanted to give you a quick heads up! Aside from that, fantastic blog!
I used to be able to find good info from your blog articles.
An outstanding share! I’ve just forwarded this onto a friend who was conducting a little homework on this. And he in fact ordered me dinner because I stumbled upon it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanks for spending time to discuss this issue here on your web site.
11 second baking soda trik
An intriguing discussion is definitely worth comment. There’s no doubt that that you need to write more on this topic, it might not be a taboo matter but generally people do not speak about these subjects. To the next! All the best!
After looking into a few of the blog posts on your web page, I really like your technique of writing a blog. I bookmarked it to my bookmark webpage list and will be checking back in the near future. Take a look at my web site too and let me know how you feel.
Pretty! This has been a really wonderful post. Thanks for providing these details.
Hi there! This blog post couldn’t be written any better! Looking at this article reminds me of my previous roommate! He continually kept preaching about this. I will send this information to him. Pretty sure he’s going to have a great read. Thanks for sharing!
There is certainly a great deal to learn about this issue. I like all the points you made.
I could not resist commenting. Very well written.
May I just say what a relief to find someone who truly understands what they’re talking about on the web. You definitely know how to bring a problem to light and make it important. A lot more people really need to read this and understand this side of the story. I was surprised that you’re not more popular because you most certainly have the gift.
Pretty! This was an incredibly wonderful post. Thank you for supplying these details.
Hi, I do believe this is a great website. I stumbledupon it 😉 I may come back yet again since I saved as a favorite it. Money and freedom is the best way to change, may you be rich and continue to help others.
Great post. I am experiencing a few of these issues as well..
I’d like to thank you for the efforts you have put in penning this website. I really hope to see the same high-grade blog posts by you later on as well. In fact, your creative writing abilities has motivated me to get my own, personal site now 😉
The very next time I read a blog, Hopefully it won’t fail me as much as this particular one. I mean, I know it was my choice to read through, nonetheless I genuinely thought you would have something interesting to talk about. All I hear is a bunch of crying about something you could fix if you weren’t too busy seeking attention.
After going over a handful of the articles on your website, I honestly like your technique of writing a blog. I saved as a favorite it to my bookmark website list and will be checking back in the near future. Take a look at my website too and let me know your opinion.
I really like reading a post that will make men and women think. Also, thanks for allowing me to comment.
Hi, I do believe this is a great blog. I stumbledupon it 😉 I’m going to revisit once again since I saved as a favorite it. Money and freedom is the best way to change, may you be rich and continue to help others.
It’s nearly impossible to find knowledgeable people about this topic, however, you seem like you know what you’re talking about! Thanks
I blog often and I really thank you for your information. This great article has truly peaked my interest. I will take a note of your website and keep checking for new information about once per week. I subscribed to your Feed too.
Your style is unique in comparison to other folks I have read stuff from. Thank you for posting when you’ve got the opportunity, Guess I’ll just book mark this blog.
Saved as a favorite, I really like your web site!
I have to thank you for the efforts you have put in penning this website. I’m hoping to view the same high-grade blog posts from you later on as well. In truth, your creative writing abilities has encouraged me to get my very own website now 😉
I really love your site.. Great colors & theme. Did you make this web site yourself? Please reply back as I’m wanting to create my own website and would love to find out where you got this from or what the theme is named. Many thanks.
I love it when individuals come together and share views. Great website, continue the good work.
I enjoy reading a post that will make men and women think. Also, many thanks for permitting me to comment.
Very good post! We will be linking to this great post on our website. Keep up the good writing.
Spot on with this write-up, I seriously believe that this site needs a great deal more attention. I’ll probably be returning to read more, thanks for the info!
Excellent web site you have here.. It’s hard to find excellent writing like yours nowadays. I honestly appreciate individuals like you! Take care!!
Oh my goodness! Amazing article dude! Many thanks, However I am encountering difficulties with your RSS. I don’t understand why I am unable to subscribe to it. Is there anybody else having identical RSS problems? Anyone that knows the answer can you kindly respond? Thanx!!
You have made some good points there. I looked on the net to learn more about the issue and found most individuals will go along with your views on this website.
This is a topic that’s close to my heart… Take care! Exactly where are your contact details though?
I was very happy to uncover this page. I wanted to thank you for your time due to this wonderful read!! I definitely appreciated every part of it and I have you book-marked to look at new things in your website.
It’s difficult to find experienced people in this particular topic, but you sound like you know what you’re talking about! Thanks
bookmarked!!, I like your web site!
Very good information. Lucky me I came across your website by chance (stumbleupon). I have saved it for later!
I enjoy reading through an article that will make men and women think. Also, thanks for allowing me to comment.
It’s hard to come by experienced people about this subject, but you seem like you know what you’re talking about! Thanks
This blog was… how do I say it? Relevant!! Finally I have found something that helped me. Thanks.
I really like reading through a post that can make men and women think. Also, many thanks for allowing me to comment.
That is a very good tip especially to those new to the blogosphere. Short but very accurate information… Appreciate your sharing this one. A must read article!
I could not refrain from commenting. Perfectly written.
Hello there! I could have sworn I’ve been to this web site before but after going through some of the articles I realized it’s new to me. Anyhow, I’m certainly pleased I discovered it and I’ll be book-marking it and checking back frequently.
Nice post. I learn something new and challenging on blogs I stumbleupon every day. It’s always exciting to read through content from other writers and use a little something from other websites.
Hi! I just want to offer you a huge thumbs up for the great information you have right here on this post. I am coming back to your blog for more soon.
Very good post. I definitely love this website. Stick with it!
Greetings! Very helpful advice within this post! It is the little changes that make the most significant changes. Thanks for sharing!
I’m impressed, I must say. Seldom do I come across a blog that’s equally educative and amusing, and without a doubt, you’ve hit the nail on the head. The issue is an issue that not enough people are speaking intelligently about. Now i’m very happy that I found this during my search for something relating to this.
I really like reading through a post that can make people think. Also, many thanks for allowing me to comment.
You’re so interesting! I do not suppose I have read a single thing like this before. So good to find another person with unique thoughts on this subject matter. Seriously.. thanks for starting this up. This web site is something that’s needed on the internet, someone with some originality.
Having read this I thought it was rather enlightening. I appreciate you taking the time and energy to put this information together. I once again find myself personally spending a lot of time both reading and commenting. But so what, it was still worth it!
I blog quite often and I truly thank you for your information. This article has truly peaked my interest. I will take a note of your blog and keep checking for new details about once a week. I subscribed to your RSS feed as well.
I could not refrain from commenting. Very well written!
I could not resist commenting. Exceptionally well written!
I want to to thank you for this very good read!! I certainly loved every bit of it. I’ve got you book-marked to look at new things you post…
Your style is unique compared to other people I have read stuff from. I appreciate you for posting when you have the opportunity, Guess I’ll just bookmark this blog.
Hello there! I just want to give you a huge thumbs up for your great info you have got here on this post. I’ll be returning to your website for more soon.
I love reading a post that can make men and women think. Also, thank you for allowing for me to comment.
You are so cool! I don’t believe I have read anything like this before. So nice to discover someone with a few original thoughts on this subject. Seriously.. thanks for starting this up. This website is one thing that is needed on the web, someone with some originality.
Pretty! This was a really wonderful post. Thanks for supplying this information.
Aw, this was a very nice post. Taking the time and actual effort to generate a great article… but what can I say… I hesitate a whole lot and don’t manage to get nearly anything done.
Everything is very open with a precise clarification of the challenges. It was really informative. Your site is very helpful. Thanks for sharing.
I wanted to thank you for this fantastic read!! I definitely enjoyed every bit of it. I have got you bookmarked to look at new stuff you post…
I’m very happy to find this site. I want to to thank you for your time for this wonderful read!! I definitely liked every little bit of it and I have you saved as a favorite to check out new things in your blog.
I’m more than happy to discover this web site. I wanted to thank you for ones time for this wonderful read!! I definitely enjoyed every little bit of it and I have you saved to fav to look at new information on your blog.
I could not resist commenting. Perfectly written!
I blog often and I really thank you for your content. Your article has really peaked my interest. I will book mark your site and keep checking for new details about once a week. I opted in for your RSS feed too.
Good post. I learn something new and challenging on blogs I stumbleupon on a daily basis. It’s always useful to read through articles from other authors and use a little something from other web sites.
Hi, I do think your blog might be having web browser compatibility issues. When I look at your site in Safari, it looks fine however, when opening in Internet Explorer, it’s got some overlapping issues. I merely wanted to give you a quick heads up! Aside from that, great blog!
After exploring a few of the articles on your blog, I honestly appreciate your way of blogging. I book marked it to my bookmark website list and will be checking back in the near future. Please visit my website as well and let me know what you think.
I’d like to thank you for the efforts you’ve put in writing this blog. I am hoping to view the same high-grade content by you in the future as well. In truth, your creative writing abilities has inspired me to get my own, personal website now 😉
I love it whenever people come together and share views. Great site, stick with it.
Can I simply just say what a relief to find somebody that truly knows what they’re discussing on the internet. You definitely understand how to bring an issue to light and make it important. More and more people should check this out and understand this side of the story. I was surprised that you’re not more popular given that you definitely have the gift.
Hi, I do believe this is an excellent blog. I stumbledupon it 😉 I may return yet again since i have book marked it. Money and freedom is the greatest way to change, may you be rich and continue to help others.
That is a very good tip particularly to those fresh to the blogosphere. Simple but very precise info… Many thanks for sharing this one. A must read post!
Howdy! I could have sworn I’ve visited your blog before but after looking at many of the articles I realized it’s new to me. Anyhow, I’m certainly delighted I came across it and I’ll be bookmarking it and checking back regularly.
Excellent article. I’m going through some of these issues as well..
That is a great tip particularly to those fresh to the blogosphere. Short but very accurate info… Thanks for sharing this one. A must read post!
Your style is so unique in comparison to other folks I’ve read stuff from. Thank you for posting when you’ve got the opportunity, Guess I’ll just bookmark this page.
You should be a part of a contest for one of the most useful blogs on the web. I am going to recommend this web site!
I’m impressed, I must say. Rarely do I come across a blog that’s both educative and interesting, and let me tell you, you’ve hit the nail on the head. The problem is something too few men and women are speaking intelligently about. I’m very happy I came across this in my search for something concerning this.
bookmarked!!, I love your web site.
Nice post. I learn something new and challenging on blogs I stumbleupon everyday. It’s always useful to read articles from other writers and practice something from other sites.
Way cool! Some very valid points! I appreciate you penning this article plus the rest of the website is also really good.
Hello there! This post could not be written any better! Going through this post reminds me of my previous roommate! He continually kept preaching about this. I will forward this information to him. Pretty sure he will have a good read. Many thanks for sharing!
Excellent post. I absolutely love this website. Keep it up!
This is a topic that is close to my heart… Cheers! Exactly where can I find the contact details for questions?
I’m impressed, I have to admit. Seldom do I encounter a blog that’s both educative and amusing, and let me tell you, you’ve hit the nail on the head. The issue is an issue that too few people are speaking intelligently about. I am very happy I found this during my hunt for something relating to this.
May I simply just say what a comfort to uncover an individual who actually understands what they’re discussing on the net. You certainly know how to bring an issue to light and make it important. More and more people must check this out and understand this side of your story. I was surprised you aren’t more popular because you definitely possess the gift.
This site was… how do you say it? Relevant!! Finally I’ve found something which helped me. Thank you.
Hello, I think your website might be having web browser compatibility problems. When I look at your web site in Safari, it looks fine however, when opening in I.E., it has some overlapping issues. I merely wanted to provide you with a quick heads up! Besides that, great blog!
That is a very good tip particularly to those fresh to the blogosphere. Brief but very precise information… Many thanks for sharing this one. A must read article.
Great info. Lucky me I discovered your website by chance (stumbleupon). I have saved as a favorite for later!
I want to to thank you for this fantastic read!! I absolutely loved every bit of it. I have you saved as a favorite to look at new stuff you post…
I love looking through an article that can make men and women think. Also, thanks for permitting me to comment.
Good write-up. I certainly appreciate this website. Thanks!
I want to to thank you for this excellent read!! I definitely enjoyed every bit of it. I have you book-marked to look at new things you post…
Hi there, I think your blog may be having internet browser compatibility problems. Whenever I look at your site in Safari, it looks fine however, when opening in Internet Explorer, it has some overlapping issues. I just wanted to give you a quick heads up! Aside from that, great site.
Good article. I am going through a few of these issues as well..
I have to thank you for the efforts you’ve put in penning this blog. I am hoping to view the same high-grade content from you later on as well. In fact, your creative writing abilities has motivated me to get my own, personal website now 😉
Good day! I could have sworn I’ve visited this website before but after browsing through a few of the articles I realized it’s new to me. Anyways, I’m definitely pleased I stumbled upon it and I’ll be book-marking it and checking back regularly.
Hi, There’s no doubt that your blog might be having web browser compatibility problems. When I look at your site in Safari, it looks fine however when opening in IE, it has some overlapping issues. I just wanted to give you a quick heads up! Aside from that, fantastic site.
I like looking through a post that will make men and women think. Also, thanks for allowing for me to comment.
There’s definately a lot to find out about this issue. I love all of the points you made.
This is a really good tip especially to those new to the blogosphere. Simple but very accurate info… Thanks for sharing this one. A must read post!
This is the right site for anybody who wants to find out about this topic. You know a whole lot its almost tough to argue with you (not that I actually would want to…HaHa). You definitely put a brand new spin on a topic that’s been discussed for a long time. Wonderful stuff, just wonderful.
Oh my goodness! Impressive article dude! Thank you so much, However I am experiencing difficulties with your RSS. I don’t understand why I am unable to join it. Is there anyone else getting identical RSS problems? Anyone that knows the solution can you kindly respond? Thanks.
Good site you have here.. It’s hard to find quality writing like yours nowadays. I really appreciate individuals like you! Take care!!
Next time I read a blog, I hope that it doesn’t fail me just as much as this particular one. I mean, Yes, it was my choice to read, nonetheless I genuinely thought you would have something helpful to talk about. All I hear is a bunch of crying about something you could fix if you were not too busy searching for attention.
I’m extremely pleased to discover this website. I wanted to thank you for your time due to this fantastic read!! I definitely enjoyed every part of it and i also have you saved to fav to check out new things in your website.
I’m extremely pleased to find this website. I want to to thank you for ones time due to this fantastic read!! I definitely really liked every little bit of it and i also have you saved to fav to look at new information in your site.
bookmarked!!, I like your blog!
Howdy! I could have sworn I’ve visited your blog before but after going through some of the posts I realized it’s new to me. Nonetheless, I’m definitely happy I found it and I’ll be bookmarking it and checking back regularly.
Hi, I do think this is a great blog. I stumbledupon it 😉 I may revisit yet again since i have saved as a favorite it. Money and freedom is the best way to change, may you be rich and continue to guide other people.
Saved as a favorite, I really like your blog.
You’ve made some really good points there. I looked on the internet to learn more about the issue and found most individuals will go along with your views on this site.
I’m impressed, I have to admit. Seldom do I come across a blog that’s both educative and amusing, and let me tell you, you’ve hit the nail on the head. The problem is something that not enough folks are speaking intelligently about. I’m very happy that I found this during my hunt for something regarding this.
After checking out a few of the blog posts on your web page, I honestly appreciate your way of blogging. I bookmarked it to my bookmark website list and will be checking back soon. Please check out my web site too and let me know what you think.
Hello! I could have sworn I’ve been to this web site before but after browsing through some of the articles I realized it’s new to me. Anyways, I’m certainly pleased I came across it and I’ll be bookmarking it and checking back often.
Next time I read a blog, Hopefully it does not disappoint me as much as this one. After all, I know it was my choice to read through, nonetheless I truly believed you’d have something useful to say. All I hear is a bunch of moaning about something you could possibly fix if you weren’t too busy searching for attention.
When I originally left a comment I seem to have clicked on the -Notify me when new comments are added- checkbox and now whenever a comment is added I recieve 4 emails with the exact same comment. There has to be a way you are able to remove me from that service? Kudos.
Everything is very open with a precise description of the issues. It was definitely informative. Your site is very useful. Many thanks for sharing.
Pretty! This has been an extremely wonderful article. Thank you for supplying this information.
Right here is the right blog for anyone who wishes to find out about this topic. You understand so much its almost hard to argue with you (not that I actually would want to…HaHa). You certainly put a fresh spin on a topic that has been discussed for years. Great stuff, just wonderful.
This site certainly has all the information I needed about this subject and didn’t know who to ask.
Good day! I could have sworn I’ve been to this site before but after going through a few of the articles I realized it’s new to me. Nonetheless, I’m definitely delighted I stumbled upon it and I’ll be book-marking it and checking back frequently!
Your style is really unique compared to other people I’ve read stuff from. Thank you for posting when you’ve got the opportunity, Guess I’ll just bookmark this page.
May I just say what a comfort to uncover someone who genuinely understands what they are talking about on the web. You certainly understand how to bring a problem to light and make it important. More people have to look at this and understand this side of your story. I can’t believe you aren’t more popular given that you certainly have the gift.
Can I simply say what a comfort to discover somebody who genuinely understands what they are discussing over the internet. You definitely realize how to bring a problem to light and make it important. More and more people must look at this and understand this side of your story. I was surprised you aren’t more popular given that you most certainly possess the gift.
I really love your website.. Very nice colors & theme. Did you develop this site yourself? Please reply back as I’m wanting to create my own personal website and want to find out where you got this from or what the theme is named. Kudos!
Good article! We will be linking to this particularly great article on our site. Keep up the good writing.
It’s hard to find knowledgeable people on this topic, however, you sound like you know what you’re talking about! Thanks
I have to thank you for the efforts you have put in writing this website. I am hoping to view the same high-grade blog posts from you later on as well. In fact, your creative writing abilities has inspired me to get my own blog now 😉
An outstanding share! I have just forwarded this onto a co-worker who has been doing a little research on this. And he actually ordered me dinner because I found it for him… lol. So allow me to reword this…. Thank YOU for the meal!! But yeah, thanx for spending time to talk about this topic here on your blog.
Your style is so unique compared to other people I’ve read stuff from. Thank you for posting when you have the opportunity, Guess I’ll just book mark this page.
That is a very good tip particularly to those fresh to the blogosphere. Simple but very precise information… Thanks for sharing this one. A must read post!
Excellent post. I absolutely appreciate this website. Stick with it!
Great post! We will be linking to this great content on our website. Keep up the good writing.
This site was… how do you say it? Relevant!! Finally I’ve found something that helped me. Thanks a lot!
Aw, this was an incredibly nice post. Finding the time and actual effort to create a very good article… but what can I say… I procrastinate a lot and never seem to get anything done.
Good post! We are linking to this particularly great content on our site. Keep up the great writing.
This website was… how do I say it? Relevant!! Finally I’ve found something which helped me. Thanks a lot!
This is a topic which is close to my heart… Cheers! Where are your contact details though?
I needed to thank you for this good read!! I definitely enjoyed every bit of it. I have you bookmarked to check out new stuff you post…
I was more than happy to uncover this great site. I wanted to thank you for ones time for this particularly fantastic read!! I definitely loved every part of it and I have you saved as a favorite to check out new stuff in your site.
I couldn’t refrain from commenting. Exceptionally well written.
This website was… how do I say it? Relevant!! Finally I’ve found something which helped me. Thanks!
Nice post. I learn something totally new and challenging on websites I stumbleupon every day. It will always be helpful to read through articles from other authors and practice a little something from their sites.
I was able to find good advice from your articles.
I must thank you for the efforts you’ve put in penning this site. I really hope to check out the same high-grade content by you in the future as well. In fact, your creative writing abilities has motivated me to get my very own blog now 😉
Aw, this was an incredibly good post. Taking a few minutes and actual effort to create a good article… but what can I say… I procrastinate a lot and don’t seem to get anything done.
Saved as a favorite, I love your web site!
Pretty! This has been an extremely wonderful post. Thank you for providing this info.
Greetings! Very helpful advice within this article! It is the little changes that will make the greatest changes. Thanks a lot for sharing!
Hello there, There’s no doubt that your site may be having web browser compatibility problems. Whenever I take a look at your site in Safari, it looks fine however, when opening in I.E., it has some overlapping issues. I just wanted to provide you with a quick heads up! Apart from that, wonderful site!
Hello there! This article couldn’t be written any better! Going through this article reminds me of my previous roommate! He always kept preaching about this. I most certainly will send this information to him. Pretty sure he will have a very good read. I appreciate you for sharing!
I wanted to thank you for this great read!! I definitely enjoyed every little bit of it. I have you book marked to check out new stuff you post…
I couldn’t refrain from commenting. Exceptionally well written!
Good web site you’ve got here.. It’s hard to find excellent writing like yours nowadays. I honestly appreciate people like you! Take care!!
Good article. I definitely love this website. Keep it up!
I have to thank you for the efforts you’ve put in penning this blog. I really hope to see the same high-grade blog posts by you later on as well. In fact, your creative writing abilities has encouraged me to get my own, personal site now 😉
An intriguing discussion is definitely worth comment. I think that you ought to write more about this subject matter, it may not be a taboo subject but usually people don’t discuss these issues. To the next! Many thanks.
The very next time I read a blog, I hope that it does not disappoint me as much as this particular one. I mean, Yes, it was my choice to read through, but I really believed you’d have something useful to say. All I hear is a bunch of complaining about something you could possibly fix if you weren’t too busy searching for attention.
Nice post. I learn something totally new and challenging on websites I stumbleupon everyday. It will always be interesting to read through articles from other authors and practice a little something from other sites.
Great post. I will be experiencing many of these issues as well..
An intriguing discussion is worth comment. There’s no doubt that that you ought to publish more about this subject matter, it may not be a taboo subject but generally folks don’t speak about these topics. To the next! Best wishes.
After I initially left a comment I seem to have clicked the -Notify me when new comments are added- checkbox and now each time a comment is added I receive 4 emails with the same comment. There has to be a way you are able to remove me from that service? Cheers.
Hello. impressive job. I did not imagine this. This is a great story. Thanks!
This website was… how do you say it? Relevant!! Finally I have found something which helped me. Thank you!
This blog was… how do I say it? Relevant!! Finally I’ve found something that helped me. Thank you!
I’m impressed, I must say. Rarely do I come across a blog that’s both educative and amusing, and let me tell you, you have hit the nail on the head. The issue is something which not enough men and women are speaking intelligently about. Now i’m very happy I came across this in my search for something regarding this.
Oh my goodness! Incredible article dude! Many thanks, However I am encountering troubles with your RSS. I don’t understand why I cannot join it. Is there anyone else having similar RSS problems? Anyone who knows the solution can you kindly respond? Thanks!!
Hello! I simply want to offer you a big thumbs up for the excellent info you have got right here on this post. I will be returning to your website for more soon.
Right here is the perfect blog for anyone who wants to find out about this topic. You know so much its almost tough to argue with you (not that I really would want to…HaHa). You certainly put a new spin on a topic which has been discussed for years. Great stuff, just great.
Everything is very open with a very clear explanation of the challenges. It was really informative. Your website is useful. Many thanks for sharing.
Having read this I believed it was rather enlightening. I appreciate you finding the time and effort to put this content together. I once again find myself personally spending way too much time both reading and posting comments. But so what, it was still worthwhile!
It’s hard to come by knowledgeable people in this particular subject, but you seem like you know what you’re talking about! Thanks
Aw, this was a really good post. Taking a few minutes and actual effort to make a great article… but what can I say… I hesitate a whole lot and never manage to get nearly anything done.
Good info. Lucky me I ran across your website by accident (stumbleupon). I’ve book marked it for later!
Great info. Lucky me I came across your site by chance (stumbleupon). I have saved it for later!
This is a topic that is close to my heart… Many thanks! Exactly where are your contact details though?
You’ve made some really good points there. I looked on the internet to find out more about the issue and found most individuals will go along with your views on this site.
bookmarked!!, I like your blog.
I used to be able to find good info from your content.
I blog frequently and I genuinely appreciate your information. This great article has really peaked my interest. I am going to book mark your website and keep checking for new information about once per week. I subscribed to your Feed as well.
I needed to thank you for this good read!! I absolutely loved every bit of it. I have got you bookmarked to look at new stuff you post…
That is a good tip especially to those new to the blogosphere. Simple but very accurate info… Thanks for sharing this one. A must read article!
Saved as a favorite, I really like your web site.
Your style is unique compared to other folks I’ve read stuff from. I appreciate you for posting when you’ve got the opportunity, Guess I will just bookmark this blog.
Hello! I simply want to give you a big thumbs up for your great information you have got right here on this post. I’ll be returning to your site for more soon.
Next time I read a blog, I hope that it won’t fail me just as much as this particular one. I mean, I know it was my choice to read through, nonetheless I truly believed you would have something helpful to say. All I hear is a bunch of moaning about something you can fix if you were not too busy looking for attention.
There is definately a great deal to know about this topic. I really like all the points you’ve made.
Hi! I could have sworn I’ve visited this site before but after browsing through a few of the posts I realized it’s new to me. Anyways, I’m certainly happy I came across it and I’ll be book-marking it and checking back regularly!
Hi there! This blog post couldn’t be written any better! Going through this article reminds me of my previous roommate! He constantly kept preaching about this. I most certainly will forward this article to him. Pretty sure he will have a great read. Thanks for sharing!
Oh my goodness! Incredible article dude! Thanks, However I am having problems with your RSS. I don’t know the reason why I am unable to subscribe to it. Is there anyone else getting identical RSS problems? Anybody who knows the solution will you kindly respond? Thanx!!
Great site you have here.. It’s difficult to find quality writing like yours these days. I seriously appreciate individuals like you! Take care!!
It’s hard to come by experienced people about this topic, however, you seem like you know what you’re talking about! Thanks
Saved as a favorite, I really like your site!
Way cool! Some very valid points! I appreciate you writing this write-up plus the rest of the site is also really good.
That is a really good tip especially to those new to the blogosphere. Brief but very precise information… Many thanks for sharing this one. A must read article.
You’ve made some good points there. I looked on the internet for additional information about the issue and found most people will go along with your views on this website.
Pretty! This was a really wonderful article. Thanks for providing these details.
Right here is the perfect web site for anyone who really wants to find out about this topic. You understand so much its almost hard to argue with you (not that I personally would want to…HaHa). You certainly put a new spin on a subject which has been discussed for decades. Great stuff, just excellent.
Hello there, I think your site could be having web browser compatibility issues. Whenever I look at your website in Safari, it looks fine but when opening in Internet Explorer, it’s got some overlapping issues. I simply wanted to give you a quick heads up! Other than that, fantastic website!
I have to thank you for the efforts you’ve put in writing this website. I really hope to see the same high-grade content from you later on as well. In truth, your creative writing abilities has motivated me to get my own, personal website now 😉
Having read this I thought it was extremely enlightening. I appreciate you spending some time and energy to put this content together. I once again find myself spending a significant amount of time both reading and posting comments. But so what, it was still worth it.
After exploring a few of the blog articles on your website, I honestly like your way of writing a blog. I saved it to my bookmark site list and will be checking back in the near future. Please visit my website as well and let me know what you think.
I really love your blog.. Great colors & theme. Did you build this web site yourself? Please reply back as I’m attempting to create my very own website and want to find out where you got this from or just what the theme is called. Thanks!
Very good article. I will be dealing with many of these issues as well..
It’s hard to find knowledgeable people for this subject, but you seem like you know what you’re talking about! Thanks
Right here is the perfect site for anybody who hopes to understand this topic. You understand so much its almost hard to argue with you (not that I personally will need to…HaHa). You certainly put a fresh spin on a subject that has been discussed for ages. Excellent stuff, just wonderful.
Having read this I believed it was extremely informative. I appreciate you finding the time and effort to put this content together. I once again find myself personally spending a significant amount of time both reading and commenting. But so what, it was still worth it!
Hi there! This post could not be written any better! Going through this article reminds me of my previous roommate! He always kept preaching about this. I will forward this article to him. Fairly certain he’s going to have a good read. Thanks for sharing!
Your style is very unique in comparison to other folks I’ve read stuff from. Thank you for posting when you’ve got the opportunity, Guess I’ll just bookmark this site.
I really love your website.. Pleasant colors & theme. Did you develop this web site yourself? Please reply back as I’m looking to create my own personal website and want to find out where you got this from or what the theme is called. Thank you.
Good web site you’ve got here.. It’s difficult to find high-quality writing like yours these days. I honestly appreciate individuals like you! Take care!!
Hello there! This post could not be written much better! Reading through this article reminds me of my previous roommate! He constantly kept talking about this. I most certainly will forward this article to him. Fairly certain he’s going to have a good read. Thanks for sharing!
Hi, I do think this is an excellent site. I stumbledupon it 😉 I may return yet again since I saved as a favorite it. Money and freedom is the best way to change, may you be rich and continue to help others.
Good web site you have got here.. It’s difficult to find good quality writing like yours nowadays. I really appreciate individuals like you! Take care!!
Very good article! We will be linking to this particularly great content on our site. Keep up the good writing.
You should be a part of a contest for one of the greatest sites on the web. I will highly recommend this site!
The next time I read a blog, Hopefully it doesn’t fail me just as much as this one. After all, Yes, it was my choice to read through, however I genuinely believed you would probably have something helpful to say. All I hear is a bunch of complaining about something that you could possibly fix if you were not too busy seeking attention.
BLUE SALT TRICK FOR MEN
Everything is very open with a precise explanation of the issues. It was really informative. Your site is very helpful. Many thanks for sharing!
Pretty! This has been an extremely wonderful article. Thanks for supplying this info.
Way cool! Some extremely valid points! I appreciate you penning this write-up and the rest of the website is also really good.
Good day! I simply would like to give you a big thumbs up for your great information you have got right here on this post. I’ll be returning to your web site for more soon.
Right here is the right web site for anybody who hopes to find out about this topic. You realize a whole lot its almost hard to argue with you (not that I personally would want to…HaHa). You certainly put a brand new spin on a topic which has been discussed for decades. Great stuff, just wonderful.
The next time I read a blog, Hopefully it does not fail me as much as this one. After all, I know it was my choice to read, however I truly thought you would probably have something interesting to say. All I hear is a bunch of complaining about something that you can fix if you were not too busy seeking attention.
After I initially left a comment I seem to have clicked the -Notify me when new comments are added- checkbox and now every time a comment is added I receive four emails with the same comment. Is there a means you are able to remove me from that service? Thanks a lot.
Howdy! I could have sworn I’ve visited this website before but after going through some of the articles I realized it’s new to me. Regardless, I’m certainly pleased I stumbled upon it and I’ll be book-marking it and checking back regularly!
I couldn’t resist commenting. Exceptionally well written!
I blog frequently and I really appreciate your content. This article has really peaked my interest. I will take a note of your blog and keep checking for new details about once a week. I opted in for your Feed as well.
Way cool! Some extremely valid points! I appreciate you penning this article and the rest of the site is very good.
Excellent blog post. I certainly love this site. Continue the good work!
Oh my goodness! Amazing article dude! Thanks, However I am having issues with your RSS. I don’t know the reason why I am unable to subscribe to it. Is there anyone else having the same RSS issues? Anyone that knows the solution will you kindly respond? Thanx!
Hi there! This post couldn’t be written much better! Going through this post reminds me of my previous roommate! He always kept talking about this. I most certainly will forward this information to him. Fairly certain he’s going to have a good read. I appreciate you for sharing!
Hello, I think your site may be having web browser compatibility problems. Whenever I look at your blog in Safari, it looks fine however, when opening in IE, it has some overlapping issues. I merely wanted to provide you with a quick heads up! Apart from that, excellent website!
Howdy! This blog post couldn’t be written any better! Going through this article reminds me of my previous roommate! He constantly kept preaching about this. I most certainly will send this article to him. Pretty sure he’s going to have a good read. Thank you for sharing!
Aw, this was an incredibly nice post. Taking the time and actual effort to make a top notch article… but what can I say… I procrastinate a lot and don’t manage to get nearly anything done.
This is a topic that is near to my heart… Many thanks! Exactly where are your contact details though?
I really like it whenever people come together and share views. Great blog, stick with it!
Way cool! Some extremely valid points! I appreciate you writing this write-up and also the rest of the site is also really good.
Uncover the details of On-Chain Crypto Mining and understand how scarcity and participation can reshape value creation. Explore how on-chain mechanics enable transparent distribution and community-led ownership.
Looking forward to reading more. Great blog post.Thanks Again. Fantastic.
Enjoyed every bit of your post.Much thanks again. Will read on…
Really enjoyed this article post.Really looking forward to read more. Fantastic.
This is one awesome post.Thanks Again. Will read on…
I’m amazed, I have to admit. Seldom do I encounter a blog that’s equally educative and amusing, and without a doubt, you have hit the nail on the head. The issue is something not enough men and women are speaking intelligently about. I’m very happy I stumbled across this during my hunt for something concerning this.
Hey there! I simply would like to give you a big thumbs up for your great info you have right here on this post. I’ll be coming back to your web site for more soon.
Have you ever considered about including a little bit more than just your articles? I mean, what you say is valuable and all. However imagine if you added some great visuals or videos to give your posts more, “pop”! Your content is excellent but with pics and videos, this website could undeniably be one of the most beneficial in its field. Awesome blog!
This site was… how do I say it? Relevant!! Finally I’ve found something that helped me. Thank you.
An intriguing discussion is worth comment. I believe that you ought to write more on this subject matter, it may not be a taboo matter but generally folks don’t speak about these topics. To the next! Kind regards.
I blog quite often and I really appreciate your information. This great article has truly peaked my interest. I will book mark your site and keep checking for new information about once a week. I subscribed to your Feed as well.
Good article. I am experiencing a few of these issues as well..
Oh my goodness! Awesome article dude! Thank you, However I am encountering difficulties with your RSS. I don’t understand the reason why I cannot subscribe to it. Is there anyone else having the same RSS issues? Anybody who knows the answer will you kindly respond? Thanx!!
Everyone loves it whenever people come together and share thoughts. Great blog, continue the good work!
After exploring a handful of the blog articles on your website, I really appreciate your way of writing a blog. I book marked it to my bookmark site list and will be checking back in the near future. Please visit my website as well and tell me how you feel.
Having read this I believed it was very enlightening. I appreciate you spending some time and effort to put this short article together. I once again find myself personally spending a lot of time both reading and posting comments. But so what, it was still worth it!
Next time I read a blog, I hope that it won’t fail me as much as this particular one. I mean, I know it was my choice to read through, nonetheless I truly believed you would have something useful to talk about. All I hear is a bunch of whining about something that you could possibly fix if you were not too busy searching for attention.
An outstanding share! I have just forwarded this onto a friend who was conducting a little research on this. And he in fact ordered me breakfast because I discovered it for him… lol. So allow me to reword this…. Thank YOU for the meal!! But yeah, thanx for spending time to talk about this matter here on your blog.
I seriously love your website.. Very nice colors & theme. Did you develop this site yourself? Please reply back as I’m wanting to create my own personal website and would love to find out where you got this from or what the theme is named. Kudos.
This blog was… how do I say it? Relevant!! Finally I have found something which helped me. Cheers!
After looking at a number of the blog posts on your web site, I really like your technique of writing a blog. I saved as a favorite it to my bookmark site list and will be checking back in the near future. Please check out my website as well and let me know how you feel.
I enjoy reading through a post that can make men and women think. Also, many thanks for allowing for me to comment.
This is a good tip particularly to those fresh to the blogosphere. Short but very precise info… Many thanks for sharing this one. A must read article!
I absolutely love your blog.. Pleasant colors & theme. Did you build this website yourself? Please reply back as I’m attempting to create my own site and would love to learn where you got this from or exactly what the theme is called. Many thanks.
Your style is very unique compared to other people I have read stuff from. Thanks for posting when you have the opportunity, Guess I will just book mark this blog.
Hey there! I simply would like to give you a huge thumbs up for your great info you have right here on this post. I am coming back to your site for more soon.
You’re so awesome! I do not believe I have read something like that before. So wonderful to discover somebody with some original thoughts on this topic. Really.. many thanks for starting this up. This site is something that is needed on the internet, someone with a bit of originality.
NEUROCEPT REVIEWS
Aw, this was a very good post. Spending some time and actual effort to create a good article… but what can I say… I procrastinate a lot and never seem to get nearly anything done.
This web site truly has all of the information I needed about this subject and didn’t know who to ask.
I want to to thank you for this wonderful read!! I absolutely loved every little bit of it. I have got you book marked to look at new stuff you post…
Everything is very open with a very clear explanation of the challenges. It was really informative. Your website is useful. Many thanks for sharing!
Hi there! I could have sworn I’ve visited this web site before but after going through some of the posts I realized it’s new to me. Regardless, I’m certainly delighted I found it and I’ll be bookmarking it and checking back often!
Good info. Lucky me I recently found your site by accident (stumbleupon). I have saved it for later!
Pretty! This was a really wonderful article. Many thanks for supplying this information.
Pretty! This has been a really wonderful post. Thanks for supplying these details.
I could not refrain from commenting. Exceptionally well written.
Hi, I believe your web site could be having browser compatibility problems. Whenever I look at your blog in Safari, it looks fine however, if opening in Internet Explorer, it has some overlapping issues. I merely wanted to give you a quick heads up! Besides that, great site.
I’m very happy to find this web site. I need to to thank you for ones time for this fantastic read!! I definitely liked every part of it and I have you book marked to look at new stuff on your site.
Good post. I learn something totally new and challenging on sites I stumbleupon everyday. It’s always exciting to read through articles from other writers and use a little something from other websites.
The next time I read a blog, I hope that it won’t disappoint me as much as this one. After all, Yes, it was my choice to read, however I really thought you would have something helpful to say. All I hear is a bunch of crying about something that you could fix if you weren’t too busy searching for attention.
Great info. Lucky me I recently found your site by accident (stumbleupon). I’ve saved it for later.
Very good info. Lucky me I found your website by chance (stumbleupon). I’ve saved it for later!
Spot on with this write-up, I honestly think this site needs a great deal more attention. I’ll probably be back again to see more, thanks for the advice.
I’m impressed, I must say. Seldom do I encounter a blog that’s both equally educative and entertaining, and without a doubt, you have hit the nail on the head. The issue is something too few folks are speaking intelligently about. I am very happy that I found this in my hunt for something regarding this.
Excellent blog you have here.. It’s difficult to find good quality writing like yours nowadays. I truly appreciate individuals like you! Take care!!
Oh my goodness! Awesome article dude! Many thanks, However I am experiencing difficulties with your RSS. I don’t know why I cannot subscribe to it. Is there anybody else having similar RSS problems? Anybody who knows the solution can you kindly respond? Thanx!
Hi, I do think this is an excellent website. I stumbledupon it 😉 I’m going to revisit yet again since i have saved as a favorite it. Money and freedom is the greatest way to change, may you be rich and continue to help others.
It’s difficult to find experienced people on this subject, but you sound like you know what you’re talking about! Thanks
Get insights into pump.fun for BNB and discover how no-code platforms are making token creation accessible to everyone.
Wonderful article! We will be linking to this great content on our site. Keep up the great writing.
I could not refrain from commenting. Very well written.
It’s hard to find knowledgeable people for this topic, but you seem like you know what you’re talking about! Thanks
Everything is very open with a clear description of the challenges. It was truly informative. Your website is very helpful. Many thanks for sharing.
I used to be able to find good advice from your blog posts.
Everything is very open with a very clear description of the issues. It was definitely informative. Your site is very useful. Thanks for sharing.
When I originally commented I seem to have clicked the -Notify me when new comments are added- checkbox and from now on whenever a comment is added I recieve four emails with the same comment. There has to be a means you are able to remove me from that service? Cheers.
Greetings! Very useful advice within this post! It is the little changes that make the largest changes. Thanks for sharing!
I truly love your website.. Very nice colors & theme. Did you make this amazing site yourself? Please reply back as I’m trying to create my own personal blog and would like to find out where you got this from or just what the theme is named. Thanks!
After looking into a handful of the articles on your blog, I truly like your technique of blogging. I added it to my bookmark webpage list and will be checking back in the near future. Please check out my web site too and let me know your opinion.
Hi, I do think this is an excellent web site. I stumbledupon it 😉 I may come back once again since i have book marked it. Money and freedom is the greatest way to change, may you be rich and continue to help others.
Your style is really unique in comparison to other folks I have read stuff from. Many thanks for posting when you have the opportunity, Guess I’ll just book mark this web site.
I truly love your site.. Pleasant colors & theme. Did you create this site yourself? Please reply back as I’m trying to create my own blog and would like to find out where you got this from or exactly what the theme is named. Thank you.
This site truly has all the info I needed concerning this subject and didn’t know who to ask.
I quite like reading a post that will make men and women think. Also, thanks for permitting me to comment.
Greetings, There’s no doubt that your blog might be having internet browser compatibility issues. When I take a look at your site in Safari, it looks fine however, if opening in IE, it’s got some overlapping issues. I just wanted to give you a quick heads up! Besides that, excellent website!
Greetings! Very useful advice within this article! It is the little changes that will make the biggest changes. Thanks for sharing!
I quite like reading through a post that can make men and women think. Also, many thanks for allowing me to comment.
This site was… how do you say it? Relevant!! Finally I have found something that helped me. Thank you.
May I simply say what a relief to uncover somebody that really understands what they are talking about on the internet. You definitely realize how to bring a problem to light and make it important. More people have to read this and understand this side of the story. It’s surprising you are not more popular because you most certainly possess the gift.
I blog quite often and I truly appreciate your information. The article has truly peaked my interest. I’m going to book mark your site and keep checking for new details about once per week. I subscribed to your Feed as well.
I couldn’t refrain from commenting. Exceptionally well written.
I was very pleased to find this web site. I want to to thank you for your time for this particularly fantastic read!! I definitely liked every part of it and I have you bookmarked to see new things in your site.
Right here is the perfect webpage for everyone who really wants to find out about this topic. You understand so much its almost tough to argue with you (not that I really would want to…HaHa). You certainly put a brand new spin on a topic that has been discussed for many years. Great stuff, just wonderful.
I really like it when individuals get together and share views. Great blog, stick with it!
Wonderful article! We will be linking to this particularly great content on our website. Keep up the good writing.
I’m amazed, I must say. Seldom do I encounter a blog that’s equally educative and engaging, and let me tell you, you have hit the nail on the head. The problem is something which not enough people are speaking intelligently about. I am very happy that I stumbled across this in my hunt for something relating to this.
Can I just say what a comfort to uncover somebody who really knows what they’re discussing over the internet. You certainly know how to bring an issue to light and make it important. A lot more people really need to read this and understand this side of your story. I can’t believe you are not more popular given that you definitely possess the gift.
You ought to be a part of a contest for one of the finest websites on the net. I am going to recommend this site!
Aw, this was an exceptionally good post. Finding the time and actual effort to generate a really good article… but what can I say… I hesitate a lot and don’t manage to get nearly anything done.
Nice post. I learn something totally new and challenging on blogs I stumbleupon on a daily basis. It will always be useful to read through content from other writers and practice a little something from other web sites.
I’d like to thank you for the efforts you’ve put in penning this website. I really hope to see the same high-grade blog posts from you later on as well. In truth, your creative writing abilities has inspired me to get my own, personal website now 😉
Everything is very open with a really clear clarification of the issues. It was really informative. Your site is very helpful. Many thanks for sharing.
An interesting discussion is worth comment. I think that you need to write more on this subject, it may not be a taboo subject but generally people do not speak about such topics. To the next! All the best.
Way cool! Some very valid points! I appreciate you penning this write-up and also the rest of the site is very good.
Spot on with this write-up, I really believe this site needs a lot more attention. I’ll probably be returning to see more, thanks for the information!
Excellent post. I will be dealing with many of these issues as well..
You ought to be a part of a contest for one of the greatest websites on the web. I am going to highly recommend this blog!
I was more than happy to discover this great site. I want to to thank you for ones time for this wonderful read!! I definitely loved every little bit of it and I have you book-marked to check out new information in your web site.
bookmarked!!, I love your web site.
This website was… how do you say it? Relevant!! Finally I have found something which helped me. Many thanks!
Greetings! Very helpful advice in this particular post! It’s the little changes that produce the most significant changes. Many thanks for sharing!
Greetings, I think your blog may be having browser compatibility issues. Whenever I take a look at your site in Safari, it looks fine but when opening in IE, it’s got some overlapping issues. I just wanted to give you a quick heads up! Other than that, wonderful blog.
I’m impressed, I have to admit. Rarely do I come across a blog that’s both educative and entertaining, and let me tell you, you’ve hit the nail on the head. The issue is something too few men and women are speaking intelligently about. Now i’m very happy I came across this in my search for something concerning this.
I like reading through an article that will make men and women think. Also, many thanks for allowing for me to comment.
An outstanding share! I’ve just forwarded this onto a friend who has been doing a little research on this. And he in fact ordered me dinner simply because I stumbled upon it for him… lol. So let me reword this…. Thanks for the meal!! But yeah, thanx for spending the time to discuss this issue here on your internet site.
Howdy! This post couldn’t be written much better! Reading through this article reminds me of my previous roommate! He constantly kept preaching about this. I am going to forward this article to him. Pretty sure he will have a great read. Many thanks for sharing!
Hi there! I just want to give you a big thumbs up for your excellent info you’ve got here on this post. I will be returning to your blog for more soon.
Good post! We will be linking to this great content on our site. Keep up the great writing.
There is certainly a lot to know about this topic. I like all of the points you made.
Howdy! I simply wish to offer you a big thumbs up for your great information you’ve got here on this post. I will be returning to your blog for more soon.
An intriguing discussion is worth comment. I think that you need to publish more on this subject matter, it may not be a taboo subject but typically folks don’t speak about these issues. To the next! Best wishes!
Your style is really unique in comparison to other folks I’ve read stuff from. I appreciate you for posting when you have the opportunity, Guess I will just bookmark this web site.
I used to be able to find good information from your blog posts.
You’re so cool! I don’t suppose I’ve read a single thing like that before. So nice to find somebody with some genuine thoughts on this topic. Really.. many thanks for starting this up. This site is one thing that is needed on the internet, someone with some originality.
Having read this I thought it was rather enlightening. I appreciate you taking the time and effort to put this article together. I once again find myself spending a significant amount of time both reading and posting comments. But so what, it was still worth it.
You made some good points there. I looked on the web for more info about the issue and found most individuals will go along with your views on this site.
Oh my goodness! Incredible article dude! Thank you, However I am having issues with your RSS. I don’t understand the reason why I cannot join it. Is there anybody getting the same RSS problems? Anyone that knows the answer can you kindly respond? Thanx.
Everything is very open with a precise explanation of the issues. It was truly informative. Your website is very useful. Thanks for sharing!