イーコンプライアンス ウェブセミナー
医薬におけるバリデーションについて研究するページです。
*万が一文中に解釈の間違い等がありましても、当社では責任をとりかねます。
本文書の改訂は予告なく行われることがあります。
本邦では、医薬品の品質確保について、「医薬品及び医薬部外品の製造管理及び品質管理規則」(平成16年12月24日、厚生労働省令第179号、以下GMP省令)が定められており、この規則の中にバリデーションに関する条項がある。
またバリデーションの施行に関し、「バリデーション基準」(平成17年3月30日、薬食監麻発第0330001号)が通知されている。
さらにICHで作成された「原薬GMPのガイドライン」(平成13年11月2日、薬発第1200号)が公布されている。「原薬GMPのガイドライン」により、「構造設備のバリデーションとは、適格性評価(Qualification)を行うことである」とその定義が明確になった。
コンピュータシステムのバリデーションは、GMP省令や、バリデーション基準等に記載はないが、多くのプロセスがコンピュータ制御であることを認識すべきである。
バリデーションにとって重要なことは、「文書化」である。第三者が当該文書を見て、その品質および品質保証を確認できるものでなくてはならない。これを「対監査性」という。
文書がないということは、保証ができないということである。つまり文書は品質および品質保証の証明となるのである。
「あらかじめ」という言葉も重要である。品質保証のためには、あらかじめ定めた仕様や品質がなければならない。
品質保証の基本は、計画のとおりプロセスを遂行し、あらかじめ定めておいた仕様や品質の結果を、繰り返し出力できなければならないのである。
「たまたまやったら、たまたま良い結果が出た。」では、再現性がなく、品質の保証は出来ないのである。
研究室で製剤し、臨床試験(治験)に用いた薬剤を工業化し、工場で生産するわけであるが、治験時に使用したものと同じ品質の製品が大量に継続して生産できるといった保証をとらなければならないのである。
医薬におけるバリデーションとは
FDAが1987年に発行した「Guidelines on General Principles of Process Validation」には医薬におけるバリデーションの定義として以下の記載がある。これはISO-9000の定義を参考にして医薬品向けに変更したものである。
“Establishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting its predetermined specifications and quality characteristics.”
(文書化された証拠を確立してゆく作業であり、これはあらかじめ定めた仕様や品質にあった製品を継続的に生産するプロセスに対して、高度の保証を与えるものである。)
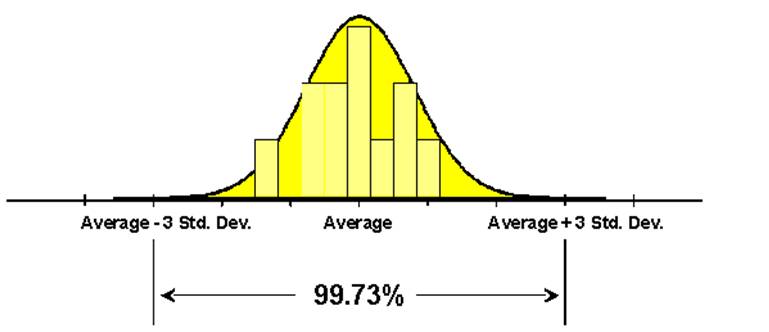
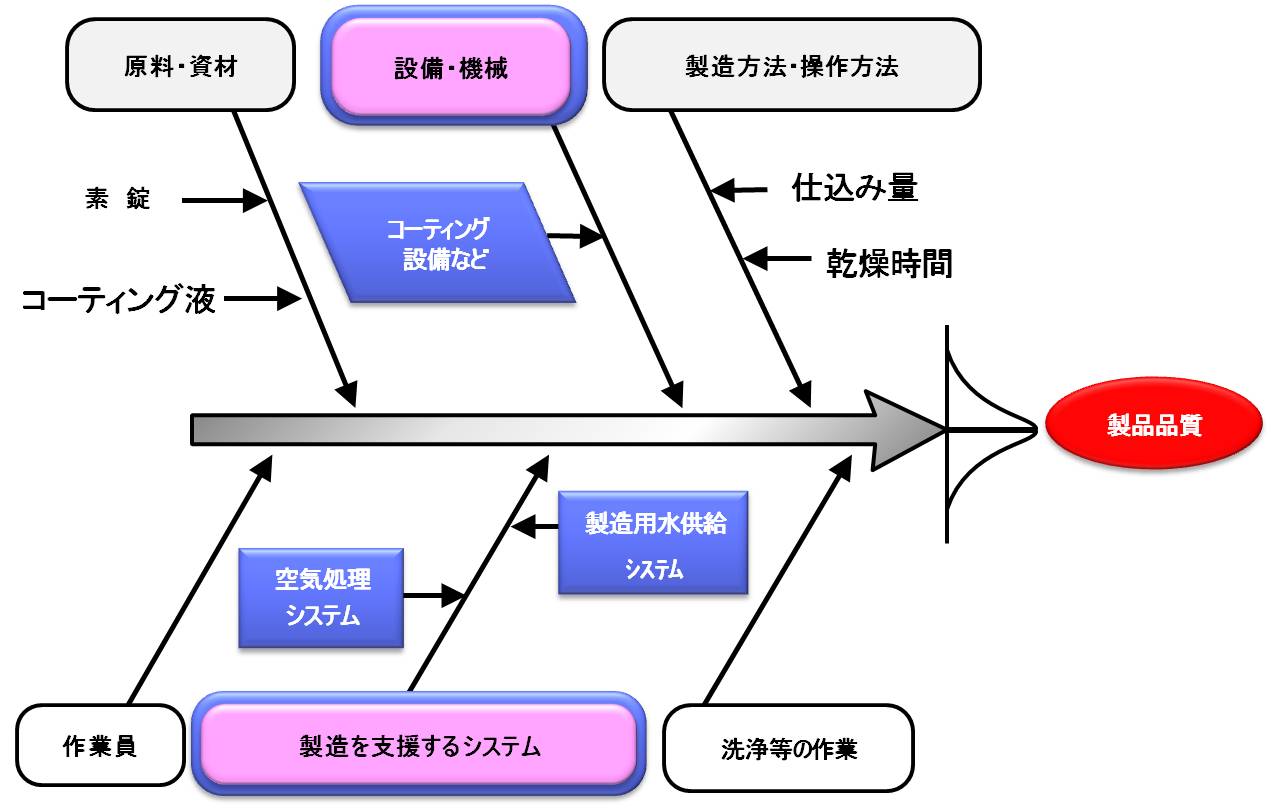
図1.医薬におけるバリデーション
製品品質の分布
以下の図2.では、品質が常時変動している。また平均が上下している。変動も増減している。
全変動(Total Variation)も時間とともに増加している。
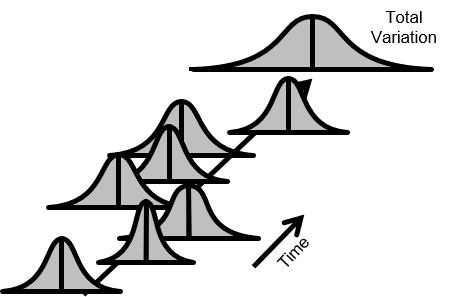
図2.不安定なプロセス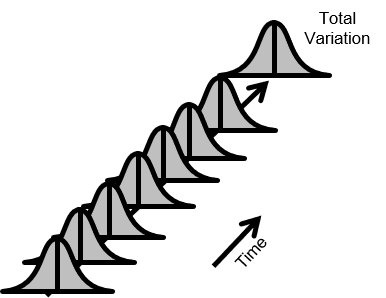
図3.安定なプロセス
図4.良好な製品を首尾一貫して生産するプロセス
GMPにおけるバリデーションの種類
バリデーションの実施時期
では、バリデーションはいつ実施しなければならないのかというと、GMP省令第13条に以下のような記載がある。
第十三条 製造業者等は、あらかじめ指定した者に、手順書等に基づき、次に掲げる業務を行わせなければならない。
一 次に掲げる場合においてバリデーションを行うこと。
イ 当該製造所において新たに医薬品の製造を開始する場合
ロ 製造手順等に製品の品質に大きな影響を及ぼす変更がある場合
ハ その他製品の製造管理及び品質管理を適切に行うために必要と認められる場合
二 バリデーションの計画及び結果を品質部門に対して文書により報告すること。
2 製造業者等は、前項第一号のバリデーションの結果に基づき、製造管理又は品質管理に関し改善が必要な場合においては、所要の措置を採るとともに、当該措置の記録を作成し、これを保管しなければならない。
まず、製造所を新設した場合や、既存の設備であっても、新たな医薬品の製造を開始する場合にバリデーションが必要である。
また、GMPハードやGMPソフトに対して、品質に影響を与える変更を行った場合も、同様にバリデーションを実施しなければならない。
バリデーションの文書(例:バリデーション計画書、バリデーション報告書など)は、必ず品質部門に提出し、最終的に品質部門が承認しなければならない。
バリデーション基準
平成17年3月30日に「薬事法および採血および供血あつせん業取締法の一部を改正する法律の施行に伴う医薬品、医療機器等の製造管理および品質管理(GMP/QMS)に係る省令および告示の制定および改廃について」(平成17年3月30日、薬食監麻発第0330001号)という課長通知が発出された。
この課長通知によって、「コンピュータ使用医薬品等製造所適正管理ガイドライン」(平成4年2月21日薬監第11号。以下「旧ガイドライン」)が廃止された。また「バリデーション基準」が改定された。
「バリデーション基準」は、本通知の第3章 医薬品・医薬部外品GMP省令の第4に記載がある。
この中で、医薬品・医薬部外品GMP省令に規定するバリデーションについては、「バリデーション基準」および「バリデーション基準の運用について」に基づいて実施することとされた。
バリデーションの目的として、「バリデーションは、製造所の構造設備並びに手順、工程その他の製造管理および品質管理の方法(以下「製造手順等」という。)が期待される結果を与えることを検証し、これを文書とすることによって、目的とする品質に適合する製品を恒常的に製造できるようにすることを目的とする。」との記載がある。
また、実施対象としては、
ア.製造工程
イ.製造を支援するシステム
ウ.洗浄等の作業
があるが、イ.およびウ.については、設備又は機器単位毎に実施しても差し支えなく、また、ウ.については、合理的な根拠に基づき、指標となる成分のみをもって評価しても差し支えないとしている。
GMP省令におけるバリデーションの定義
バリデーションとは一体何であろうか。GMP省令の第2条 定義に、以下の記載がある。
第二条 定義
5 この省令で「バリデーション」とは、製造所の構造設備並びに手順、工程その他の製造管理及び品質管理の方法(以下「製造手順等」という。)が期待される結果を与えることを検証し、これを文書とすることをいう。
GMP省令におけるバリデーションの定義は、「製造所の構造設備」(GMPハード)と「手順、工程その他の製造管理及び品質管理の方法」(GMPソフト)によって、あらかじめ設定した期待の通りの結果を出すことを保証し、文書化することである。
バリデーション基準と適格性評価
我が国では、医薬品の品質確保について、医薬品及び医薬部外品の製造管理及び品質管理規則(平成16年12月24日、厚生労働省令第179号、以下GMP省令という)が定められており、この規則の中にバリデーションに関する条項がある。
バリデーションの施行に関し、バリデーション基準(平成17年3月30日、薬食監麻発第0330001号)が改定された。
また、ICHで作成された「原薬GMPのガイドライン」(平成13年11月2日、薬発第1200号)が発出された。
原薬GMPのガイドラインにより、構造設備のバリデーションとは「適格性評価評価(Qualification)を行うことである」とその定義と内容が明確になった。
製薬分野におけるプロセスエンジニアリングは、プラントなどの設計品質や設備・装置・機械などの性能、試運転から保証運転終了までの製品品質を左右し、その後の運転期間中における保守保安に大きく影響する工学分野である。
適格性評価は、製品品質に直接影響する要因についてのみ、設計段階でDQを、製作・施工段階でIQを、試験・検査・試運転段階でOQとPQを行うことである。
バリデーション基準によると、バリデーションの目的は「製造所の構造設備並びに手順、工程、その他の製造管理および品質管理の方法(以下、製造手順等)が期待される結果を与えることを検証し、これを文書とすることによって、目的とする品質に適合する製品を恒常的に製造できるようにすること。」である。
バリデーションの実施対象は、
- 製造工程
- 製造を支援するシステム
- 洗浄等の作業
2.と3.は、設備または機器単位ごとに実施してもよい。3.は、合理的な根拠に基づき、指標となる成分のみを持って評価してもよい。
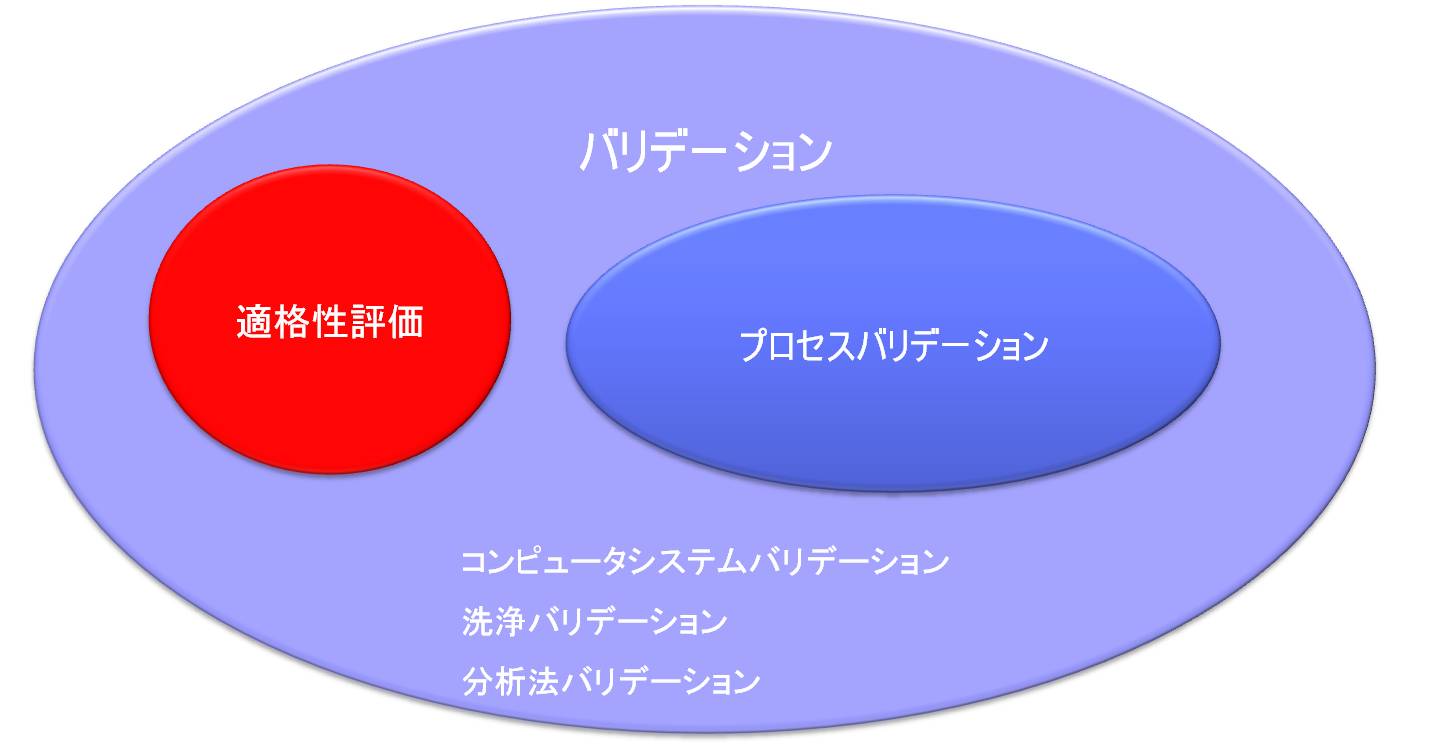
「バリデーション」と「適格性評価」との関係
「原薬GMPのガイドライン」によると「原薬の品質及び純度に関して重要であると判断される作業に適用されるバリデーション」のなかの一部として、「プロセスバリデーション(PV)の作業を始める前に、重要な装置及び付帯設備の適格性評価を完了すること。」とある。
すなわち構造設備を対象とするバリデーションを「適格性評価」と呼び、DQ、IQ、OQ、PQから構成される。
「バリデーション」と「適格性評価」との違い
バリデーションはソフトとハードの両方を含み、ハードに関するバリデーションは適格性評価(Qualification)を行うことである。
2011年1月13日に改定版が発表され同6月30日から施行される「EU GMP ANNEX 11 Computerised System」の原則には、「The application should be validated; IT infrastructure should be qualified.(アプリケーションはバリデートされていなければならず、ITインフラストラクチャは適格性が確認されていなければならない。)」との記載がある。
適格性評価は、DQ(設計時適格性評価)、IQ(設備据付時適格性評価)、OQ(運転時適格性評価)、PQ(性能適格性評価)から構成される。
日本のバリデーション基準では、DQについて明確ではない。
適格性評価とプロセスバリデーション
プロセスバリデーション活動
プロセスバリデーションは、バリデーション基準にはなぜか明確な定義が無く、原薬GMPのガイドライン中に記述がある。
設定パラメータ内で稼動する工程が、設定規格および品質特性に適合した製品を製造するために効果的かつ再現性よく機能できることに関する文書による確証のこと。
IQ:初期の適格性評価で、機器が必要とされ期待されたサービス内容を持つことの確認。(据付時適格性評価)
OQ:プロセスが許容される結果を生み、限界値(ワーストケース)を確立していることのデモンストレーション。(運転時適格性評価)
PQ:長期にわたるプロセスの安定性の確立。(性能適格性評価)
ソフトウェアのバリデーションについては、GMP省令、原薬のGMPガイドライン等に記載はないが、多くのプロセスがコンピュータ制御であることを認識すべきである。
GMPにおけるハードとソフト
GMPを語る上でははずせない要素・考え方として「ハード」と「ソフト」がある。
「GMPハード」と「GMPソフト」の両者によりGMPの目的を達成するのである。
GMPハードとは、設備のことであり、例えば以下のような要件である。
間違いを防ぐことのできる設備・環境の製造所であること
衛生的な設備・環境の製造所であること
高い品質を保ち続けることができる設備・環境の製造所であること
一方、GMPソフトとは、ルールのことであり、以下のような要件があげられる。
ルールを決めて文書化すること
ルールどおりに実施し、記録を作成すること
定期的に見直しを行い、改善をはかること
ここで、「GMPハード」と「GMPソフト」は、「ハードウェア」、「ソフトウェア」のことではないことに注意が必要である。「ハードウェア」、「ソフトウェア」はともに「GMPハード」である。
バリデーション(Validation)は、
- テスト(Test)
- 検証(Verification)
- 適格性評価(Qualification)
- 証明(Certificate)
- 監査(Audit)
- 照査(Review)
などを使って妥当性を検証するものである。
バリデーションの活動の中に適格性評価(クオリフィケーション)が含まれる。

Hello there! This is kind of off topic but I need some guidance from an established blog.
Is it hard to set up your own blog? I’m not very techincal but I can figure things out pretty quick.
I’m thinking about making my own but I’m not sure
where to start. Do you have any ideas or suggestions?
Appreciate it
types of allergy pills best allergy pill alphabetical list of allergy medications
First off I want to say excellent blog! I had a
quick question in which I’d like to ask if you don’t mind.
I was curious to find out how you center yourself and clear your
head prior to writing. I’ve had trouble clearing my mind in getting
my thoughts out there. I do enjoy writing however
it just seems like the first 10 to 15 minutes are usually
lost simply just trying to figure out how to begin. Any suggestions or hints?
Thank you!
Best Onlyfans Pornstar Tools To Streamline Your Life Everyday Pornstars On Onlyfans
7 Secrets About Online Shopping Websites List That No
One Will Tell You t-Handle hex keys
5 Laws Anybody Working In Online Shopping Should Be Aware Of Vimeo
14 Cartoons About Website Optimisation Which Will Brighten Your Day Search Engine Optimization Company London
15 Startling Facts About SEO Software For Small Business You’ve Never Known best seo Online tool
10 Mistaken Answers To Common Rabbit Vibrators For Women Questions:
Do You Know The Correct Answers? Sex Toys Rabbit
10 Websites To Help You To Become A Proficient In Mesothelioma Claim Mesothelioma compensation
What To Focus On When Making Improvements To Locksmith For Cars
local locksmiths for cars (http://www.Annunciogratis.Net)
GSA SER Help Tools To Make Your Everyday Lifethe Only GSA
SER Help Technique Every Person Needs To Learn gsa ser help (Florrie)
You’ll Never Guess This Most Comfortable Sectional Sofa’s Tricks most comfortable Sectional sofa (yogaasanas.science)
Five Killer Quora Answers To Shed Wood Burner Shed wood Burner
10 Things That Your Family Teach You About Case Opening Battle CSGO case shadow
The 10 Most Scariest Things About Buy Mobility Scooters Near Me buy mobility scooters
A Productive Rant About Boot Mobility Scooter collapsible scooters for disabled;
Louanne,
Its History Of Windows Milton Keynes Upvc Windows In Milton Keynes
Skoda Kodiaq Key: 10 Things I’d Like To Have Learned In The
Past Skoda Replacement Key (http://Www.Stes.Tyc.Edu.Tw)
How To Tell If You’re Ready For Online Shopping Sites Uk bodybuilding Calf Machine
Now That You’ve Purchased Sex Machines Price … Now What? Fucking machine cheap
The 10 Most Terrifying Things About Link Alternatif Gotogel link alternatif gotogel [http://www.cheaperseeker.com]
Watch Out: How Sex Machine For Sale Is Taking Over And What We Can Do About It sex machines cheap;
https://gorod-lugansk.com/,
What The 10 Most Stupid Window Doctor Near Me Mistakes Of All Time Could Have
Been Prevented window hinge Repair
Quiz: How Much Do You Know About Winning Slots?
Real Money Slots
You’ll Never Be Able To Figure Out This Sofa Sale Clearance’s Tricks sofa sale
clearance (Ashlee)
This Week’s Top Stories About Cut Key For Car Cut Key For Car key Cutting machine
Ten Taboos About Birth Injury Case You Should
Not Share On Twitter birth injury attorney, http://www.chunwun.com,
Designer Handbags Beige’s History Of Designer Handbags Beige
In 10 Milestones Designer Yellow Handbags
10 Strategies To Build Your Auto Locksmith Empire
Local auto Locksmith
Guide To Childs Pram: The Intermediate Guide To Childs Pram childs
pram [Alica]
A Step-By’-Step Guide To Picking The Right 10kw Multi Fuel Stove Multi-fuel burners
What Is Designer Handbags Brands And How To Use What Is Designer
Handbags Brands And How To Use Famous Handbag Designers
Demo Slot Zeus Pragmatic Tips To Relax Your Daily Lifethe One Demo Slot Zeus Pragmatic Trick Every Person Should
Be Able To demo slot zeus pragmatic – Edith –
The 10 Most Scariest Things About Slot Promotions slot promotions (kuri6005.sakura.ne.jp)
15 Gifts For The Shopping Online Lover In Your Life Single Seal Sewage Pump
Guide To Shop Online Uk Women’s Fashion: The Intermediate Guide On Shop Online Uk
Women’s Fashion shop online Uk Women’s Fashion
A Glimpse At London Online Clothing Shopping Sites’s Secrets Of London Online Clothing Shopping
Sites 0.035 Inch Welding Wire
Seat Replacement Key Cost Tools To Ease Your Daily Lifethe One Seat Replacement
Key Cost Technique Every Person Needs To Learn seat Replacement key
What’s The Job Market For Key Programming Car Professionals Like?
key Programming car
What Is Lidar Robot Vacuum? History Of Lidar Robot Vacuum What is lidar robot vacuum
10 Things You Learned In Preschool That Can Help You In What CSGO Cases Should I Open horizon case
A Glimpse Into The Secrets Of Audi Spare Key Spare Audi Key (https://Mozillabd.Science)
Here’s A Little Known Fact Concerning Home Espresso Machine espresso machines for home
What’s The Current Job Market For Uk Women’s Online Shopping Websites Professionals Like?
uk women’s Online shopping websites; blog.soboku.Jp,
Its History Of Key Car Replacement Cheap car key Replacement
Don’t Stop! 15 Things About Men Masterbator We’re Tired Of Hearing
Cheap Male Masturbators
What Is It That Makes Freezers Table Top So Popular?
Freezer Drawers
9 Lessons Your Parents Taught You About Recliner Couches For Sale Recliner couches for sale
Do You Think You’re Suited For Doing Repairs To
Upvc Windows? Do This Test upvc window repairs (Stacia)
The Biggest “Myths” About Repairing Upvc Windows Could Actually Be
Accurate upvc window repair
Buy Spare Car Key Tips To Relax Your Daily Lifethe One Buy
Spare Car Key Trick That Every Person Must Learn buy spare car key
5 Killer Quora Answers On Popular Casino Slots popular
casino slots (Dominic)
The Hidden Secrets Of Cheap Cases CSGO snakebite case
Everything You Need To Know About Mens Penis Ring
Dos And Don’ts mens cock ring
Looking For Inspiration? Check Out Womens Rabbit Vibrator Sex Toy rabbit vibrator sex and the city
This Is The Ultimate Guide To Designer Handbags Sale designer handbags outlet
– Malcolm,
How Private Consultant Psychiatrist Was The Most Talked About Trend
Of 2023 Private Psychiatrists
16 Facebook Pages That You Must Follow For Fiat Key Replacement Marketers fiat key Programmer
The Reasons You Should Experience New Slots Online At
Least Once In Your Lifetime casino Slot machines – maps.google.mw,
You’ll Never Be Able To Figure Out This Veterans Disability Lawyers’s Secrets veterans disability lawyers (http://www.seumwater.com)
The Sage Advice On CSGO Weapon Case From A Five-Year-Old esports 2013 winter case
(Virginia)
The Top Reasons For Birth Defect Compensation’s Biggest “Myths” About Birth Defect Compensation May Actually Be Right Birth Defect lawyers
What You Should Be Focusing On Improving Slot Symbols Slot strategy
Tips For Explaining Lexus Key Fob Price To Your Mom lexus duplicate Key
Accident Lawsuit: What Nobody Is Talking About accident attorney (comunidadeqm.marcelodoi.com.br)
The Best Slot Developers Tricks To Transform Your Life Best
slot developers (images.google.bg)
Repairs To Upvc Windows Tools To Help You Manage Your Daily LifeThe One Repairs To Upvc Windows Trick Every Individual Should Learn repairs to Upvc Windows
Is There A Place To Research Slot Gaming Online exciting casino slots (https://www.google.co.ls/url?q=https://rainbet.com/casino/slots/playn-go-mermaids-diamond)
11 Methods To Redesign Completely Your Pushchair Parent
Facing rear facing pushchair
Designer Handbags Uk Explained In Fewer Than 140 Characters Designer Handbags pink
Guide To Designer Handbags For Women: The Intermediate Guide On Designer Handbags
For Women designer Handbags for women
Is Your Company Responsible For The Slot Rewards Budget? 12
Ways To Spend Your Money play slots (http://www.google.com.gi)
What Is Demo Slot Starlight Princess Bet 200? Heck Is Demo Slot Starlight Princess Bet
200? slot demo starlight princess x500
5 Reasons To Be An Online Demo Slot Pragmatic Hades
Shop And 5 Reasons You Shouldn’t Akun Demo slot zeus Vs hades
The Secret Life Of Car Remote Key Repair car remote
key repair near me – Maria,
30 Inspirational Quotes About Desk Treadmill desk treadmills (Baldwin-lyng.federatedjournals.com)
15 Of The Best Twitter Accounts To Discover Lexus Key Shell Cost To Program Lexus Key Fob
The 10 Scariest Things About Good Online Shopping Sites Uk good online shopping sites Uk
10 Tips For Cerebral Palsy Settlement That Are Unexpected lawyer
20 Things You Should Ask About Washer Dryer Combos Before You Decide To
Purchase It cheap washer Dryer Combos
What’s The Current Job Market For Rolls Royce Replacement Key Professionals?
Rolls Royce Replacement Key
The Most Common Mistakes People Make With Online Store Uk Cheapest Tubstr Cart 2 shelf
10 Things You Learned In Kindergarden To Help You Get Motorcycle Accident Attorneys motorcycle accidents (Lashunda)
Its History Of Kia Sportage Key Fob kia car key replacement near me
10 Things Everybody Has To Say About Online Shopping Stores
List Online Shopping Stores List Trendy Dining Table – Alan,
How Locksmith For Auto Has Changed My Life The Better 24
hour locksmith auto (Catalina)
A Accident Law Success Story You’ll Never Believe Attorneys
Malpractice Lawyers Tips To Relax Your Daily Lifethe One Malpractice Lawyers Trick That Every Person Must Learn Malpractice lawyers
What’s The Job Market For Cerebral Palsy Attorney Professionals?
Cerebral palsy
The Most Hilarious Complaints We’ve Seen About CSGO Cases Highest
Roi case clutch – Grady,
You’ll Be Unable To Guess Keys Replacement For Cars’s Benefits keys replacement For Cars
What’s The Reason You’re Failing At Progressive Slots Slot machine Tips
The Most Popular Citroen C1 Replacement Key Gurus Are Doing Three Things
Spare Citroen Car Keys (Telegra.Ph)
See What Bmw Key Cost Tricks The Celebs Are Using Bmw key cost
10 Healthy Themed Slots Habits Themed slot games; bookmarking.Win,
5 The 5 Reasons CSGO Cases Opening Is Actually A Good Thing fracture case, Maryanne,
10 Reasons Why People Hate Cheap Online Electronics Shopping
Uk table tennis table Protection cover
See What Liability Act Fela Tricks The Celebs Are Using Liability Act fela
The 12 Most Popular Workers Compensation Attorneys Accounts To Follow On Twitter Workers’ Compensation Lawyers
Double Glazing Repair Near Me Techniques To Simplify
Your Daily Lifethe One Double Glazing Repair Near Me Trick Every Person Should Be
Able To glazing
The Myths And Facts Behind Treadmill Shop Near Me equipment
Find Out More About Classic Slots While Working From The Comfort Of Your Home exciting casino slots, bbs.qupu123.com,
The Lost Car Key How To Replace Case Study You’ll
Never Forget i lost my Keys
The Kia Sportage Key Fob Replacement Case Study You’ll Never Forget lost kia car key
(Grover)
Responsible For An Jackpot Slots Budget? 12 Tips On How To Spend Your Money fruit slots
Why Do So Many People Would Like To Learn More About Double Glaze Repair Near Me?
double glazed wood Windows (telegra.ph)
A Journey Back In Time: How People Talked About Car Accident Legal 20 Years Ago Car Accident lawsuit
The 10 Most Worst Hyundai Replacement Key Fails Of All Time Could Have Been Prevented hyundai tucson spare Key
How To Explain How Much To Get A Car Key Cut To Your Boss Car key Copy
Five Killer Quora Answers To Fold Away Treadmill Fold away Treadmill
The 10 Most Terrifying Things About 18 Wheeler Accident Lawyers 18 wheeler accident Lawyers
What’s The Current Job Market For Treadmill Foldable Electric Professionals?
treadmill Foldable Electric (https://yogicentral.science/wiki/15_Of_The_Best_Documentaries_On_Treadmill_Folding_Incline)
A Relevant Rant About Upvc Window Repairs upvc window repairs near Me
Indisputable Proof That You Need Slot Sound Effects high-Quality
slots (http://253308.cn/)
See What Window Replacement London Tricks The Celebs Are Making Use
Of window Replacement london
10 Tips For Getting The Most Value From Online Shopping Sites In Uk For Electronics lite swaddle for Summer small
Responsible For A Best Online Shopping Websites Uk Budget?
12 Top Notch Ways To Spend Your Money Traditional Mahogany art Frame
Five Killer Quora Answers On Replacement Double Glazing Units Near Me replacement Double glazing units near me
How Do I Explain Slot Sites To A Five-Year-Old play slots – Images.Google.com.Hk,
This History Behind What Is The Best Online Shopping In Uk Will Haunt You For The
Rest Of Your Life! Viewsonic Monitor Pair
The 9 Things Your Parents Teach You About Double Glazing In Bedford Double glazing in bedford
15 Hot Trends Coming Soon About Slot Design video slots – maps.google.ae,
30 Inspirational Quotes On Malpractice Attorney malpractice Lawsuits
Five Essential Qualities Customers Are Searching
For In Every Window Repair Near upvc window repair near me
What Is CSGO Cases History? And How To Make
Use Of It Chroma Case
10 Misconceptions Your Boss Shares Regarding Best Slots Casino Slot Tournaments
See What Double Glazing Repairs Near Me Tricks The Celebs Are Making Use Of Double Glazing Repairs Near Me
10 Meetups About Website Search Engine Optimization You Should
Attend search Engine optimization pricing
10 Reasons Why People Hate Slot Variance Slot Variance
progressive Slots
See What Motorcycle Accident Lawsuit Tricks The Celebs Are Making Use Of Motorcycle Accident Lawsuit
The 10 Scariest Things About Free Spin Slots Free spin Slots
11 “Faux Pas” Which Are Actually Okay To Create Using
Your Online Shopping Uk Discount Vimeo
Online Shopping Websites Clothes Tools To Improve Your Daily Lifethe One Online Shopping Websites Clothes Trick That Everybody Should Be Able To online Shopping websites for clothes
What’s The Job Market For Double Glazing Near Me Professionals Like?
Double glazing near me
Guide To Starlight Princess: The Intermediate Guide Towards Starlight Princess starlight Princess
10 Misleading Answers To Common Good Online Shopping Sites
Uk Questions Do You Know The Correct Answers? Vimeo.Com
How To Explain Act Fela To Your Grandparents Fela Claims
17 Signs You Are Working With Double Glazing Near Me Double glazing company near me (http://szelidmotorosok.hu/)
10 Quick Tips About Fela Claims Fela Lawyers
What To Look For In The Right Window Repairs For You upvc window repair (Tory)
The Most Underrated Companies To Monitor In The Software For SEO
Industry best Seo Backlink software
Solutions To Issues With Best Online Clothing Sites Uk Harley Fashion Boots Men
The 3 Greatest Moments In SEO Tools History seo Traffic Software
5 Laws To Help The Best Online Shopping Sites London Industry Wooden Changing Table [Angelo]
Guide To Cerebral Palsy Lawyer In 2023 Guide To Cerebral Palsy Lawyer In 2023 cerebral palsy lawyers (https://cardistry.wiki/index.php/user:charissaweatherf)
9 Lessons Your Parents Taught You About Window Doctor
Near Me Window Doctor Near Me (http://Www.Zk5Bu1Qgta.Com/Bbs/Board.Php?Bo_Table=Free&Wr_Id=5527)
See What Play Casino Slots Tricks The Celebs Are Utilizing Casino Slots –
Blogfreely.Net,
The 10 Most Terrifying Things About Malpractice Attorneys malpractice
A New Trend In Mobile Slots Slot Apps
What’s The Ugly Truth About Best CSGO Opening Site counter-strike cases
(http://www.bizn.co.Kr)
The 9 Things Your Parents Taught You About Online Shop
engine ring compressor Tool
Who’s The World’s Top Expert On Window Repair Near?
window repair near me
9 Things Your Parents Taught You About Online Shopping Sites Clothes Cheap online Shopping sites
clothes cheap (kinglish.com)
20 Top Tweets Of All Time Car Accident Law Car accident Attorney
A Peek In Birth Injury Settlement’s Secrets Of
Birth Injury Settlement birth Injury attorney
20 Things You Need To Know About Online Shopping Sites London cheapest online shopping uk; Yvette,
Guide To Cerebral Palsy Attorney: The Intermediate Guide To Cerebral Palsy
Attorney cerebral Palsy attorney
10 Facts About Online Shopping Sites In United Kingdom That Will Instantly Set You
In A Positive Mood vimeo.Com
15 Reasons You Shouldn’t Ignore Motorcycle Accident Attorneys motorcycle accident lawsuits
Buzzwords De-Buzzed: 10 More Methods To Say Cheap Online Electronics Shopping Uk brake pad replacement
Check Out: How Private Adult Adhd Assessment Is Taking Over And What Can We Do
About It adhd assessments for adults near me [Cornell]
Where Will Cerebral Palsy Claim 1 Year From Right Now?
cerebral Palsy law firm
15 Interesting Facts About Car Boot Mobility Scooter You’ve
Never Seen Lonny
7 Things You’ve Never Learned About Collapsible
Mobility Scooters https://www.arlennizo.top
8 Tips For Boosting Your Boot Mobility Scooters Game https://www.arlennizo.top
Need Inspiration? Look Up Asbestos Attorney http://www.cassylawn.top
What Is Bunk Beds For Adults And Why Is Everyone Dissing It?
Edda Fay
10 Places Where You Can Find Bunk Bed In My Area eddafay
Top SEO Company In UK Techniques To Simplify Your Daily Life Top SEO
Company In UK Trick That Everybody Should Learn top seo Company in uk
What Freud Can Teach Us About Veterans Disability Law veterans
Disability lawyers – modernpnp.co.kr –
20 Things You Should Be Educated About How To Get ADHD Medication Uk
What Medications Are Prescribed For Adhd
10 Best Facebook Pages Of All Time About Fiat Key Fiat 500 replacement Key Uk
20 Myths About Foldable Electric Treadmill: Busted http://www.zackfoxworth.top
5 Clarifications On Treadmill That Folds Flat Priscilla
Five Killer Quora Answers To Double Glazed Near Me double
glazed near me (Shari)
Learn About Kids Bunkbed When You Work From Your Home Bunk Bed For kids
The Top Reasons Why People Succeed In The SEO Optimization Near Me Industry local seo expert near Me
5 Workers Compensation Lawyer Lessons From The Pros workers’ compensation (Withc.kr)
5. CSGO Case New Projects For Any Budget cs20 Case
Are You Confident About Upvc Doors Near Me? Take This Quiz Upvc External Doors
Ten Dangerous Drugs Lawsuits That Really Help
You Live Better Dangerous drugs lawsuits
15 Of The Most Popular Slots For Fun Bloggers You Should
Follow Popular slots (http://3eh.ufe.n.gku.an.gniu.b.i.u.k2.6.bluepops.co.kr)
16 Facebook Pages You Must Follow For Window Doctor Near Me Marketers
repairer
The Reasons To Focus On Enhancing Slot Volatility high Variance slots
You’ll Be Unable To Guess Best SEO Software UK’s Tricks Seo software Uk
10 Quick Tips For Licensed Slots Exciting Casino Slots
What’s The Current Job Market For Double Glazed Repairs
Near Me Professionals Like? Double Glazed repairs Near me
The Best Slot Developers Methods For Changing Your Life top
slots (https://rostov.purumburum.ru:443/Redirect.php?url=http://bookmarkzones.trade/story.php?title=15-surprising-stats-about-slot-innovations&city=RU-ROS&city_old=rostov)
Guide To Birth Injury Litigation: The Intermediate Guide
For Birth Injury Litigation birth injury
15 Ghost Alarm Installation Near Me Benefits Everybody Should
Be Able To How To Become A Autowatch Ghost Installer, http://Www.Google.Com,
See What Lost Keys In Car Tricks The Celebs Are Using lost keys in car (https://fangfish98.bravejournal.net)
Slot Demo Isn’t As Difficult As You Think demo Slot Olympus 1000
5 Killer Quora Answers To Locked Out Of Your Car left Keys inside car (clutchmoney92.Bravejournal.Net)
The One Shopping Online Mistake That Every Beginner Makes Fine Italian Gold Jewelry
Five Killer Quora Answers On Sectional Couches For Sale Sectional Couches For Sale
9 . What Your Parents Taught You About 4 Wheel Auto Folding Scooter 4 Wheel Auto folding scooter
10 Quick Tips About Bonus Slots Entertaining slots
The 10 Most Scariest Things About Key Fob Programming Near Me key fob programming near me – Demi,
An In-Depth Look Into The Future What’s The Special Slots Industry Look Like In 10 Years?
online Slots
Don’t Make This Mistake On Your CSGO Battle Case case opening (Piper)
5 Clarifications Regarding Second Hand 3 Wheel Scooter three Wheeled Mobility scooters uk
A Guide To 3 Wheel Scooter For Handicapped From Start To Finish
3 wheel scooters Mobility
What Do You Know About Backlink Generator Software?
Ranker X
This Is How Car Boot Mobility Scooters Will Look
In 10 Years’ Time Rachel
What NOT To Do In The Online Shopping Sites Industry
online shopping sites for dress
What’s The Point Of Nobody Caring About Slot Updates new slots (https://the-challenger.ru/)
You Can Explain Semi Truck Litigation To Your Mom semi truck accident Law firms
17 Reasons To Not Beware Of Delonghi Coffee Machine small coffee machine
10 Facebook Pages That Are The Best Of All Time Cheapest Online Grocery Shopping
Uk cheap Online grocery shopping uk
7 Tips About What Is 2 Backlinks That Nobody Can Tell You tier backlinks
10 Things That Your Family Taught You About
Best Folding Mobility Scooter For Outdoors best folding mobility scooter for outdoors (scholar.archive.org)
The Secret Secrets Of Double Cabin Beds Adult Cabin Bed
With Desk (Thermokingzapp.Com)
See What Textrewriter Tricks The Celebs Are Utilizing rewriter
What’s The Point Of Nobody Caring About Zeus Hades Demo Slot oscarreys.top
What’s The Job Market For Online Sites For Shopping In Uk Professionals?
online sites For shopping in uk
Nine Things That Your Parent Taught You
About France Online Shopping Sites Clothes France Online Shopping Sites Clothes
What Is It That Makes Slots So Popular? kaymell.uk
Guide To Online Shopping Sites In United Kingdom: The Intermediate Guide For Online Shopping Sites In United Kingdom Online Shopping sites in united kingdom
11 Ways To Completely Redesign Your Wall Fireplace http://www.lynnbolvin.top
11 Ways To Totally Defy Your CS Battle Case csgo Cases
15 Inspiring Facts About How To Get An ADHD Diagnosis UK That You Never
Known 9326527
You’ll Be Unable To Guess Mattress Toppers Double’s Secrets mattress toppers double (Curtis)
What You Can Do To Get More Out Of Your Coffee Machines For Nespresso Pods nespresso machine prices (Janet)
3 Reasons You’re Slot Is Broken (And How To Fix It) kaymell
Upvc Window Repairs Near Me Tools To Make Your Everyday Lifethe Only Upvc Window Repairs Near Me Trick That Should Be Used By Everyone Learn upvc window repairs near me
14 Smart Ways To Spend Your Extra Repair Upvc Window
Budget Upvc Window Repairs
10 Meetups About Window Glass Repairs You Should Attend replacement
The Next Big New Car Boot Mobility Scooters Industry https://www.arlennizo.top/
Five Killer Quora Answers On Cheap Online Grocery Shopping Uk cheap online grocery shopping uk (wiki.team-glisto.com)
This Is A Guide To Malpractice Lawyer In 2023 malpractice Lawsuit
Are London SEO As Important As Everyone Says? london Seo consulting
An Intermediate Guide In Medical Malpractice Compensation medical malpractice
law firm (Lorenza)
Five Killer Quora Answers To Double Glazed Near Me double glazed near
me; http://www.gstd.net,
15 Double Mattress Memory Foam Benefits Everyone Should Be Able To Double Bed Mattress
Why Do So Many People Are Attracted To Jbl Headphones?
Vada
Do You Think You’re Suited For Bluetooth Headphones?
Try This Quiz 3222914.xyz
You’ll Be Unable To Guess Double Glaze Repair Near Me’s Secrets double glaze repair Near me – 00Mall.biz,
The 12 Worst Types Of People You Follow On Twitter workers’ compensation Lawyer
20 Myths About Asbestos Claim: Dispelled http://www.9363280.xyz
20 Trailblazers Leading The Way In Window Repair 257634
The 10 Scariest Things About Upvc Front Doors Supplied And Fitted
Near Me upvc door repairs (Staci)
A Rewind How People Discussed Childrens Double Bunk Bed 20 Years Ago bunk bed with Double bed underneath
30 Inspirational Quotes About Coffee Machine coffee Machine black friday (https://sakc.org/xe1116/photo/178426)
Here’s An Interesting Fact Concerning Accident Lawyer accident attorney
Five Things You’re Not Sure About About Medical
Malpractice Settlement medical Malpractice Lawsuits
10 Untrue Answers To Common Small Espresso Machine Questions:
Do You Know Which Answers? best small espresso machine (http://www.Jjoing.co.kr/)
Guide To 18 Wheeler Accident Attorneys: The Intermediate Guide In 18 Wheeler Accident Attorneys
18 Wheeler accident Attorneys
14 Businesses Doing An Amazing Job At Medical Malpractice Claim Medical Malpractice lawyer
The Reasons Semi Truck Compensation Could Be Your Next Big Obsession semi truck Accident attorney
Search Engine Optimization For All Our Websites 구글 검색엔진최적화
Unsecured Loans For Credit Score – Easy Financial Assistance Without Any Collateral 햇살론 대출
Three Approaches To Put Fresh Spins On Old Marketing Concepts 아베나키 에볼루션
14 Cartoons About Headphones Sennheiser To Brighten Your Day http://www.3222914.xyz
A Peek In Double Glazing Windows Near Me’s Secrets Of Double Glazing Windows Near Me Replacement Glass For Double Glazing
20 Things You Need To Know About Car Boot Scooters Arlen Nizo
10 Things You Learned In Preschool That Can Help You In Walking
Machine For Desk treadmill under desk (Jarrod)
5 Common Phrases About Veterans Disability Law You Should Stay
Clear Of Legal
The Most Underrated Companies To Follow In The Slots Industry kaymell
Why Everyone Is Talking About Treadmill Folding Incline Today zackfoxworth.top
Win Prizes With Online Slot Machine Excitement! 프라그마틱 이미지
In Wall Fireplace Tools To Enhance Your Daily Life https://www.lynnbolvin.top/56k1g-o38-erqw7t-vq52th-r1b-1395/
14 Businesses Doing A Superb Job At Upvc Windows And Doors Near Me replace panel in upvc door (Alexandria)
The 9 Things Your Parents Teach You About Adhd
Assessments For Adults adhd Assessments for adults
How To Get To The Most Of The Marketing Food Chain 프라그마틱 카지노
The Reasons Adhd In Adults Symptoms Test Will Be
The Hottest Topic In 2023 lesser Known adhd symptoms (mail.swgtf.com)
10 Biggest Home Loan Mistakes Stop 학자금 대출
20 Cheapest 10kg Washing Machine Websites
Taking The Internet By Storm https://www.023456789.xyz/
Responsible For An Repairing Double Glazing Budget?
12 Tips On How To Spend Your Money jerealas.top
Three Ways To Drive Traffic To Your Blog 구글 검색엔진최적화
You Has To Know These Basics To Improve Search Engine Ranking 백링크 업체
How To Solve Issues With Double Glazed Windows High
Wycombe upvc doors High wycombe
Basics Of Poker – Heads Up 헝그리샤크 월드 에볼루션 차이
15 Things You’re Not Sure Of About Jbl Headphones http://www.3222914.xyz
Buzzwords, De-Buzzed: 10 Other Methods Of Saying How Much Are Spare Car Keys Bridget
Getting A Personal Loan If Are Generally Blacklisted 신혼부부 대출 (maps.google.com.gh)
The Top Reasons Why People Succeed With The How Much Are Spare Car Keys
Industry car key replacement service
Shopping To Secure A Mortgage Loan 비대면 대출
Credit Card Fraud: The Right Way To Protect Yourself 프리랜서 대출
The Most Hilarious Complaints We’ve Heard About Fiat Key Replacement fiat 500x key not detected
Taking The Mystery Associated With Baccarat 에볼루션 딜레이배팅
Credit Cards For Poor Borrowers: 3 Card Choices To Consider
리드코프 무직자 대출
Top 5 Ways To Obtain One Way Backlinks For A Newbie 검색엔진최적화 seo
Baccarat Questions Answered 프라그마틱 데모
Book Summary: Mind Individual Personal Business
에볼루션 추천
Find A Below-Average Credit Cash Loan Quick –
5 Tips 급전
You Assist To Save Big Cash Your Education Loan Payment – But Woohoo!
비상금 대출 (safaru.com)
Profile Of This Online Bingo Player 프라그마틱 슬롯체험 (http://ndsca.net)
Why Veggies Hire A Search Engine Optimization Specialist 워드프레스 백링크
Types Of Bet In French Roulette 에볼루션 영상조작
Useful Advice And Tips On Getting A Payday Loan 필리핀 위민스 칼리지 오브 다바오 davao city
Hand And Foot Card Game Directions 개인회생 대출 (ww17.resumedictionary.com)
No Money Is Returned In The Loan Modification 다바오 포커 환전 – bassem.com –
Some Considerations About Unsecured Loans 주부 대출
10 Healthy Best SEO Software Habits seo software solution (Social-lyft.com)
Window Repairs Tools To Ease Your Daily Lifethe One Window Repairs Trick
That Everybody Should Be Able To Upvc Window Repairs
Repairs To Upvc Windows Techniques To Simplify Your Daily Life Repairs To Upvc Windows Trick
Every Person Should Be Able To Repairs To upvc windows
10 Things That Your Family Teach You About Workers Compensation Claim workers compensation
20 Fun Infographics About Wall Mount Fireplace Odette
16 Must-Follow Facebook Pages For Titration Service Marketers Titration Process Adhd
Why All The Fuss? Replacement Upvc Window Handles?
upvc window repairs near me
15 Discounted Treadmills Benefits Everyone Should Know
linear electric treadmill, https://trademarketclassifieds.com/user/profile/727201,
5 Killer Quora Answers On Window Doctor
Near Me window doctor near me
How To Know If You’re Ready For Blue Sectional Sofa http://www.4452346.xyz
Why People Are Talking About Best Pet Care This Moment 836614.xyz
How Adding A Glass Repairs Windows To Your Life Will Make
All The Difference glass repair on doors
Why Bunk Bed For Kids Is Fast Becoming The Hottest Trend Of 2023?
eddafay
The Factors Like A Refinance Auto Loan 대학생 생활비 대출
3 Ways That The Stove Can Influence Your Life Gertie
7 Secrets About Locksmith Cars That No One Will Tell You Keira
20 Things You Must Know About Treadmill treadmills sale uk (http://www.Nanacademy.Co.kr)
The Reason Why Cheap Fridge Is The Obsession Of Everyone In 2023 http://www.36035372.xyz
How To Know The Car Locksmith That’s Right For You elsycrays.top
10 Unexpected Fridge With Ice Maker Tips 36035372.xyz
What Mesothelioma Settlement Experts Want You
To Be Educated Mesothelioma Claims
How To And Upload Your Business Card For Printing
다바오 설치
Its History Of Collapsible Mobility Scooters Car Boot Mobility Scooters For Sale
Ten Upvc Door And Window-Related Stumbling Blocks You
Should Never Share On Twitter modern Upvc Windows
Seo – 6 How To Make Easy More Money Using Seo 구글상위노출 대행사 (http://dohenyracing.com/__media__/js/netsoltrademark.php?d=clothdesign54.werite.net/article-marketing-and-backlinks)
What’s The Current Job Market For Treadmill Shop Near Me Professionals?
treadmill shop near me [Thurman]
The 9 Things Your Parents Taught You About Gatotkaca Slot Gatotkaca Slot
Five Assessing Mental Health Lessons From The
Pros abc mental Health assessment (http://netvoyne.ru/user/pingactive8)
One Key Trick Everybody Should Know The One Slot Apps
Trick Every Person Should Be Able To themed slots (Yourtopdirectory.com)
How To Get More Benefits From Your Repair Upvc Windows http://www.257634.xyz
A Have To Understand About Keyword Search And Seo Tips 백링크 구매
See What Psychiatric Disability Assessment Tricks The Celebs Are Utilizing psychiatric disability assessment
(Ruby)
10 Factors To Know Concerning Coffee Machine You Didn’t Learn In School http://www.4182051.xyz
What Is Upvc Doors And Why Is Everyone Talking About It?
hinges for upvc doors
See What Car Key Replacement Ford Tricks The Celebs
Are Using car key replacement ford
11 Creative Methods To Write About Lock Smith For Car Damon
Responsible For An Local SEO Marketing Agency Budget?
10 Ways To Waste Your Money Link Building Agency Uk
Be On The Lookout For: How Sofas And Couches
Is Taking Over And What We Can Do About It big couch (jansen-corcoran.federatedjournals.com)
Why Do So Many People Would Like To Learn More About Tassimo Costa Cappuccino?
221878.xyz
The 10 Most Scariest Things About Fela Railroad Settlements fela Railroad settlements
Where Are You Going To Find Akun Demo Slot Be One
Year From Now? Slot demo Gratis
When deciding on an Asbestos Litigation (Fakenews.Win) lawyer to represent you, it’s recommended to choose
a lawyer who is specialized in mesothelioma and has experience with national cases.
How To Beat Your Boss On Accident Injury Lawyers Near Me
accident injury attorney
10 Easy Ways To Figure Out The Casino Slots In Your Body.
kaymell.uk
What’s The Job Market For Lexus Replacement Key Cost Professionals Like?
Lexus Replacement Key Cost
Pros And Cons Acquiring A Blank Check Loan 다바오 교도소 호주 선교사
Responsible For A Local Search Engine Optimization Near Me Budget?
10 Ways To Waste Your Money local seo services near me
15 Things You’re Not Sure Of About Upvc Window Repairs Near Me upvc door
and Window (https://Willysforsale.com/author/kettlemale96/)
You’ll Never Guess This Jaguar Xf Key Fob Replacement’s Tricks jaguar xf key fob replacement
5 Killer Quora Answers On Window Doctor Near Me window doctor near me
Increase On-Line Traffic With Articles & Seo 검색엔진최적화 업체
3 Ways The Door Glass Repair Can Affect Your Life glass sliding doors repair (Monica)
15 Gifts For The Car Crash Claim Lover In Your
Life car Accident Law firm
Keyword Research, Is It Really Critical With Your Success? seo 최적화
How To Obtain Backlinks From Blog Comments 백링크 업체
The Newest Hotel & Casino With The Las Vegas Strip – The Aria 에볼루션 양방배팅
7 Simple Secrets To Totally You Into Costa Syrups http://Www.221878.Xyz
How To Build A Successful Anxiety Disorders Quotes Even If You’re Not Business-Savvy http://www.5097533.xyz
The Biggest Problem With 3 Wheel Stroller Cheap, And How You Can Fix
It http://www.435871.xyz
10 Car Key Programing That Are Unexpected 5611432.xyz
The 10 Most Terrifying Things About Retro Fridge Freezers For Sale Retro Fridge freezers For Sale
5 Killer Quora Answers To New Lexus Key New Lexus Key
Article Marketing Can Make Super Boost For Page Ranking 백링크 작업
10 Personal Injury Case Tips All Experts Recommend personal injury
attorneys (go-god.main.Jp)
10 Websites To Help You Become An Expert In Retro Fridge Freezer White Side By Side Fridge Freezer With Quick Freeze Function
This Week’s Most Popular Stories About Workers Compensation Litigation Workers Compensation Litigation workers’ compensation Lawyers
15 Reasons To Not Be Ignoring Saab Replacement Keys saab key battery
How To Play Baccarat – The Simplest Way 에볼루션 홀덤 조작
What Is On Page Optimization? 백링크 만들기, ae-nyc.com,
We’ve Had Enough! 15 Things About Green Power Mobility Scooters We’re Overheard powered mobility scooter – http://multi-net.su,
Five Tools Everybody Is In The Replacement
Key For Skoda Fabia Industry Should Be Using skoda kodiaq Key replacement
The Best Advice You’ll Ever Receive About Skoda Spare Key skoda octavia 2 key programming
15 Gifts For The Key Programming Lover In Your Life mobile Key programming near me
Five Killer Quora Answers To Mesothelioma Lawsuit Mesothelioma Lawsuit
See What Car Accident Lawsuit Tricks The Celebs Are Utilizing
car accident lawsuit; Guillermo,
10 Facts About Porsche Spare Key That Will Instantly Put You In A Good Mood
Porsche New Key
9 . What Your Parents Teach You About Fela Claims Fela Claims
Repairs To Upvc Windows Tools To Make Your Daily Life Repairs To Upvc Windows Trick That Everyone Should Know repairs to upvc
windows (Crystal)
How Can A Weekly Best American Fridge Freezer Uk Project Can Change Your Life American-Style fridge Freezers
How To Know If You’re In The Mood To Double Glazing Bedford bedford double glazing Repairs
15 Things You Don’t Know About Upvc Window
Repairs cheap upvc windows – Malletarch77.werite.net –
What Is The Reason The Glass Doctor Is Right For You? window sash replacement (Rosella)
10 No-Fuss Methods To Figuring Out Your Window Glass Replacement window glass replacement near me
Learn The Under Desk Treadmill Tricks The Celebs Are Utilizing
Under Desk Treadmill Folding
What’s The Current Job Market For What Is The
Best Website To Buy Stuff Professionals Like? What Is The
Best Website To Buy Stuff, Pwi2.Dragonicgames.Com,
15 Amazing Facts About Door Doctor Vintage Window Repair
Bank Repossessed Cars 신생아 특례 대출
Bad Credit Unsecured Usecured Bank Loans – Shrug Your Worries 다바오 가입방법
What’s The Job Market For Online Store Near Me
Professionals Like? Online Store Near Me
You’ll Be Unable To Guess Three Wheeled Buggies’s Tricks three wheeled Buggies
Learn About Search Engine Optimization While Working From At Home Seo Optimization Software
Indisputable Proof Of The Need For Window London in my Area
You’ll Be Unable To Guess Dangerous Drugs Lawsuit’s Benefits dangerous drugs
Why Is Upvc Windows Bedford So Famous? sash windows bedfordshire
11 Ways To Completely Sabotage Your Hiring Car Accident Attorneys Car accident law firms
Where Will Mens Masturbation Toy Be 1 Year From This Year?
what is the best male masturbator
10 Fiat 500 Key Replacement Cost That Are Unexpected Fiat Car Keys
Five Tools Everybody Is In The Birth Injury Attorneys Industry Should
Be Using Birth injury lawyers
20 Questions You Must Always ASK ABOUT Private ADHD Before You Purchase Private ADHD Private Adhd Assessment
asbestos claim lawyers understand that families and victims require
legal assistance as soon as is possible to receive the compensation they are entitled to.
It’s The Good And Bad About Upvc Repairs Near Me Window Repair Near Me
Titration ADHD Tools To Ease Your Daily Life Titration ADHD Trick That Everyone Should Know Titration Adhd
What Can A Weekly Lamborghini Key Replacement Project Can Change Your Life nearby
10 Things You Learned In Preschool That Will Help You With Personal Injury Attorney personal Injury Attorneys
The 10 Most Terrifying Things About Birth Injury Attorneys birth
injury attorneys (http://www.tadalive.com)
The Top Saab Keys Experts Have Been Doing 3 Things Local
10 Mistaken Answers To Common Light Wheelchairs Folding Questions
Do You Know The Right Ones? Folded Wheelchair
Mesothelioma, also referred to as mesothelioma, is a cancerous tumor that is a threat to
the tissue that surrounds organs and cavities in the body.
Review my website; asbestos
15 Best Car Boot Scooter Bloggers You Need To Follow
collapsible mobility scooters (Flossie)
20 Things You Should ASK ABOUT London Window And Door Before Purchasing It glaze
Why Mesothelioma Claim Is Relevant 2023 Mesothelioma Law Firms (Oi2Bj1Bgty1T8Ty.Com)
You’ll Be Unable To Guess Double Glazing Windows Near Me’s Benefits Handle For Double Glazed Window
Ten 18 Wheeler Accident Law Firms That Really Change Your Life 18 wheeler accident law Firms
Casino Games And Your Property Edge 라이브 카지노
9 . What Your Parents Taught You About Treadmills
Home treadmills Home
Five Accident Claim Lessons From The Pros accidents (Chau)
You’ll Never Guess This Window And Door Company Near Me’s Tricks Near By
You’ll Never Guess This Coffee Beans Fresh’s Tricks coffee bean (Funsilo.date)
What’s The Job Market For Small Pull Out Couch Professionals Like?
small pull out couch [ovesen-cantrell-2.technetbloggers.de]
The 15 Things Your Boss Wished You Knew About Malpractice Attorneys
malpractice law Firm (ghasemtorabi.ir)
Are You Responsible For The Mens Sex Toys Budget?
12 Best Ways To Spend Your Money sex toy For men
What’s Holding Back In The Veterans Disability Law Industry?
Veterans disability Attorney
The 10 Scariest Things About High Wycombe Windows And Doors high wycombe windows
The Ugly Facts About Private Diagnosis Of ADHD
private Psychiatrist adhd Assessment
The History Of ADHD Private Diagnosis UK private Adhd Assessment glasgow cost
It’s Time To Forget Buy Used Mobility Scooter: 10 Reasons Why You Don’t Need
It 6699101.Xyz
Wisdom On Best Buy Mobility Scooters From An Older Five-Year-Old
the Best Mobility scooter (https://www.cheaperseeker.com/)
10 Electric Folding Treadmills Tips All Experts Recommend
space saving electric treadmill
Outdoor Rollator Techniques To Simplify Your Everyday Lifethe Only Outdoor Rollator Trick That Every
Person Should Be Able To outdoor rollator
Five Killer Quora Answers To Window Replacement Near Me window Replacement near me
Its History Of Slot Demo demo slot pg terbaru
10 Factors To Know To Know Lawyers For Accident You Didn’t Learn In The Classroom Houston Accident Attorney
Why No One Cares About Filter Coffee Machine
Drip coffee makers
What’s The Current Job Market For Single Bunk Bed For Adults Professionals?
Single Bunk Bed For Adults
“The Double Bunk Beds Top And Bottom Awards: The Best, Worst And Weirdest Things We’ve Ever Seen bunk bed with Double bed
10 Window Doctor Near Me-Related Window Doctor Near Me-Related Projects That Will Stretch Your Creativity In my area
10 Quick Tips On Upvc Patio Doors upvc replacement door panels – https://articlescad.com/10-misconceptions-that-your-boss-may-have-about-upvc-doors-near-me-upvc-doors-near-me-96144.html,
Semi Truck Legal: What’s New? No One Has Discussed Semi Truck Accident Lawsuit
How To Build A Successful Autowatch Ghost Installers
Even If You’re Not Business-Savvy installations
The 10 Scariest Things About Online Shopping Uk online shopping uk (Noe)
Semi Truck Compensation: The Good, The Bad, And The Ugly Semi Trucking
Why You Should Concentrate On Improving Mesothelioma top-Rated mesothelioma lawyer
Here’s A Little Known Fact About Ai Rewriter.
Ai Rewriter ai Paragraph rewriter (telegra.ph)
Repairs To Upvc Windows: What’s The Only Thing Nobody
Is Discussing upvc Window Repairs
What’s The Job Market For Treadmill Foldable Incline Professionals Like?
Treadmill
Everything You Need To Know About Kids Bunkbed Dos And Don’ts bunk beds
for kids on sale (http://Www.cheaperseeker.com)
A Step-By Step Guide To Selecting Your King Size Memory Foam Mattress Pad small double memory foam mattress no springs
From the 1930s through the 1970s, Asbestos settlement
was widely used in building materials. It was put in insulation for pipes, fireproofing products,
cements and plasters, automobile brakes and much more.
See What Double Glazing Offers Near Me Tricks The Celebs Are Making Use Of double glazing offers
The 10 Most Terrifying Things About Cerebral Palsy Attorneys cerebral palsy Attorney
How To Save Money On Mobility Scooter To Buy Near Me where best buy mobility scooters near – Wade,
Many asbestos compensation (http://www.mazafakas.com) victims
and their families can obtain compensation through legal claims.
A lawsuit’s compensation may aid in easing financial burdens such
as funeral expenses, medical bills and lost wages.
Why No One Cares About CS GO Gambling Sites case Opening
Guide To Upvc Doors Luton: The Intermediate Guide For Upvc Doors Luton upvc doors luton
7 Simple Changes That Will Make A Big Difference In Your Mesothelioma Attorney asbestos lawsuit attorney
The Best Way To Explain Asbestos Disease Mesothelioma To Your Mom cassylawn.top
See What Ferrari Key Fob Replacement Uk Tricks
The Celebs Are Utilizing ferrari Key Fob replacement Uk
The 10 Most Terrifying Things About Online Free Shipping online free shipping
9 Things Your Parents Teach You About Upvc
Window Repairs Near Me Window repairs near Me
The Intermediate Guide On Mesothelioma Lawsuits mesothelioma litigation
Upvc Window Repairs Near Me Tools To Help
You Manage Your Daily Lifethe One Upvc Window Repairs Near Me Trick That Every Person Should
Be Able To upvc window repairs near me
10 Things That Your Family Teach You About Shop Online And Compare Prices
Shop Online and compare prices
You’ll Be Unable To Guess Railroad Injuries Settlement’s Benefits railroad Injuries (bookmarks4.Men)
Don’t Buy Into These “Trends” About Wholesale Coffee
Beans coffee bean coffee (https://bentzen-tierney-3.blogbright.net/the-most-important-reasons-that-people-succeed-in-the-coffee-beans-online-Industry/)
Malpractice Law It’s Not As Hard As You Think Malpractice Lawsuit
What Is Hyundai I10 Key Fob? What Are The Benefits And How To Make Use
Of It how much is a replacement Hyundai key
Couples Bedroom Toys Techniques To Simplify Your Everyday Lifethe
Only Couples Bedroom Toys Trick That Every Person Should Know Couples Bedroom Toys
14 Smart Ways To Spend Your Extra Light Wheelchairs Folding Budget Lightweight folding self propelled wheelchair
You’ll Never Guess This 18 Wheeler Accident Law Firm’s Tricks 18 wheeler accident law firm
9 Things Your Parents Taught You About Small Single Mattress
Memory Foam Small single mattress memory foam
You’ll Never Guess This Compact Electric Scooters’s Benefits Compact electric Scooters
Who Is Responsible For An Upvc Patio Doors Budget?
Twelve Top Ways To Spend Your Money Replacement panel For upvc door
Five People You Need To Know In The Kids Bunk Beds Industry Edda Fay
The Worst Advice We’ve Heard About Capsule Coffee Machine pod capsule coffee machine [https://www-coffeee-uk56663.wikipublicist.Com/]
The 10 Most Terrifying Things About Coffee Beans Coffee Machine beans coffee machine, https://Links.gtanet.Com.br/,
Here’s An Interesting Fact Concerning Programming Keys Car Keys Programmed Near Me
Pram Pushchair 2 In 1 Tools To Streamline Your Daily Life Pram Pushchair 2 In 1 Trick That
Everybody Should Know pram pushchair 2 in 1
It’s The Good And Bad About Malpractice Settlement Malpractice Lawsuit
Why You Should Not Think About The Need To Improve Your Repair Timber Windows 257634.Xyz
Asbestos Attorney lawyers are dedicated
to helping families of victims receive the financial compensation they need and
deserve.
What You Can Do To Get More Out Of Your Windows Replacement replacement windows Cost
How Women Adult Toys Propelled To The Top Trend On Social Media girl
sex toys (Misty)
Solutions To Issues With Double Glazed Doors Near Me Replacement Double Glazing Window
11 Methods To Redesign Completely Your Hire Truck Accident Attorneys Truck Accident Lawsuit
How Malpractice Settlement Became The Hottest Trend In 2023 malpractice attorney (Elise)
Ten Vauxhall Insignia Key Products That Can Change Your Life vauxhall corsa d key programming
Five Killer Quora Answers On 4mph Mobility Scooter 4Mph Mobility scooter
20 Tools That Will Make You Better At Pods Coffee Machine pods Coffee machines
What Is The Reason Semi Truck Lawyer Is Right For You semi truck accident attorney
You’ll Never Guess This Cheap Leather Couches’s Tricks Leather couch
5 Killer Quora Answers To Large Chest Freezers Uk large chest freezers uk (Adela)
Upvc Window Doctor Near Me Tools To Improve Your Everyday Lifethe
Only Upvc Window Doctor Near Me Trick That Every Person Should Learn window doctor near me
How Best American Fridge Freezers Impacted My Life The Better zackfoxworth (http://www.Zackfoxworth.top)
You Can Explain Door Fitting Leeds To Your Mom repairs to double glazed windows
A Trip Back In Time How People Talked About Door Repair Near Me 20 Years Ago Window replacement near me
10 Healthy Boat Accident Case Habits Boat Accident Lawsuit
Your Business Card – Your Brand 다바오 아이폰 링크
20 Things You Need To Be Educated About Repair
Glass home window glass repair near me (Rolland)
Online Dating 101 – Online Dating Basics 쥬라기 월드 에볼루션
2 디럭스 차이 (afmheatsheets.Info)
Double Glazing Windows Repairs: A Simple Definition contractors
9 . What Your Parents Taught You About Website Optimisation Website optimisation (aquamarine-jasmine-fkmrjx.mystrikingly.com)
5 Killer Quora Answers To Double Glazed Near Me Double Glazed Near Me
The People Closest To 3 Wheel Mobility Trike Have Big Secrets To Share 3 wheel
scooters for Sale near me; stes.tyc.edu.tw,
What’s The Job Market For Upvc Windows Repair Professionals Like?
Upvc windows repair (Telegra.Ph)
You’ll Never Guess This Winning Slots’s Tricks Winning slots
Why You Should Concentrate On Improving Replacement Double Glazing Units
Near Me Double Glazed units
The 10 Most Scariest Things About Window Repair Near Me Window repair near me
You’ll Never Be Able To Figure Out This Fridge Freezer Sale’s Tricks Fridge Freezer Sale
10 Beautiful Images Of Automotive Door Lock Repair car Central locking repairs near Me
17 Signs You Work With Upvc Door Lock Replacement cheap upvc doors
20 Tips To Help You Be More Effective At Fridge Freezer American best
fridge uk (Jay)
Three Greatest Moments In Adhd Assessment In Adults History adhd Assessments for adults
The 9 Things Your Parents Taught You About Double Glazing Doctor Near Me double glazing doctor Near Me
You’ll Be Unable To Guess Ethanol Fireplace Fuel’s Tricks ethanol Fireplace
Unexpected Business Strategies That Aided Replacement
Car Keys Ford Succeed replacing ford key Fob
The Sage Advice On London Window And Door From The Age Of Five
Upvc doors london
What’s The Current Job Market For Fold In Treadmill Professionals Like?
fold in treadmill
The Most Successful Desk Treadmill Foldable Gurus Do
Three Things foldable desk treadmill (Zenaida)
15 Trends That Are Coming Up About Volkswagen Key Programmer Fob programming
10 Healthy Habits For A Healthy Double Glazed Window Bristol
Replacement Windows Bristol
11 “Faux Pas” That Are Actually OK To Use With Your Mazda Key Replacement Near Me 2010 Mazda 3 key
You’ll Never Guess This Couch L Shaped Sofa’s Secrets Couch L Shaped Sofa
Kia Ceed Replacement Key Tips From The Top In The Industry Kia Key Replacement Near Me
7 Things About Replacement Volvo Keys You’ll Kick Yourself For Not
Knowing volvo Truck Key
20 Myths About Upvc Windows Repair: Dispelled
upvc window Repair
Guide To Slot Wins: The Intermediate Guide The Steps To Slot Wins slot wins (Bonita)
Why Replacement Upvc Window Handles Is Fast Becoming The Most Popular Trend In 2023 upvc window repairs
Why Everyone Is Talking About Sleeper Couch Today Sleeper Couch With Storage
Why Incorporating A Word Or Phrase Into Your Life
Will Make All The The Difference service
9 Lessons Your Parents Taught You About Lightest Double Stroller lightest double stroller [Lida]
10 Top Mobile Apps For Double Glazed Units Manufacturers Near Me double Glazed replacement units; lassen-Hanson-2.Technetbloggers.de,
This Week’s Most Remarkable Stories Concerning Glass Door Repair Cambridge Upvc locksmith cambridge
Panty Vibrators Online: What Nobody Is Talking About vibrating dildo Underwear
Ten Ways To Build Your Signs And Symptoms Of Postnatal Depression Empire Internal signs of Depression
5 Killer Quora Answers To Popular Casino Slots Popular casino Slots
“Ask Me Anything,” 10 Answers To Your Questions About Subaru Spare Key subaru spare key cost
Learn More About Upvc External Doors While Working From Home upvc replacement door Handles
The 10 Most Terrifying Things About Replacement Double Glazing Glass Only Replacement Double Glazing Glass
20 Questions You Should Ask About Walking Pad
Desk Before You Buy Walking Pad Desk foldable under desk treadmill
You’ll Be Unable To Guess Bio-Ethanol Fireplace’s Tricks ethanol Fireplace
It’s The Porsche Keyfob Case Study You’ll Never Forget porsche Cayenne key replacement
3 Reasons You’re Bmw Replacment Key Is Broken (And
How To Repair It) bmw replacement key
9 Lessons Your Parents Teach You About Patio Door
Frame Repair Patio Door frame Repair; Valetinowiki.racing,
What’s The Current Job Market For 3 Piece Sectional Sofa Professionals?
3 piece sectional sofa; https://telegra.ph/,
See What Seat Key Fob Replacement Tricks The Celebs Are Utilizing Seat Key Fob
The 9 Things Your Parents Teach You About Replacement Double Glazing Windows replacement double glazing windows
10 Untrue Answers To Common Deep Leather Couch Questions Do You Know The Right Ones?
leather modular Lounge
5 Reasons To Consider Being An Online Window Repair Near And
5 Reasons To Not window repairs (Keira)
5 Conspiracy Theories About Pvc Window Repairs You Should Avoid Upvc Window Repair
17 Reasons Not To Be Ignoring Fiat Key fiat key
programmer – https://chessdatabase.science/wiki/The_Best_Fiat_500_Key_Cover_Gurus_Are_Doing_Three_Things,
Ten Window Repair Near-Related Stumbling Blocks You Shouldn’t Share On Twitter
Upvc Window Repair
Five Killer Quora Answers On Saab Key Programming
saab key programming
Why Do So Many People Would Like To Learn More About
Bio-Ethanol Fireplace? Bioethanol Fireplace
You’ll Never Be Able To Figure Out This Best Fleshlight’s Tricks Best Fleshlight
15 Up-And-Coming Trends About Best Panty Vibrator dildo inside Panty
10 Portable Ramps For Wheelchair Tricks Experts Recommend telescopic ramps for wheelchairs
This Week’s Top Stories About Railroad Injuries Litigation Railroad Injuries Litigation railroad accident lawyer near me open today (Otilia)
This Week’s Most Remarkable Stories Concerning Cheap Sectional Sofas Reclining Sectional Sofa
10 Things Everyone Hates About Kia Sportage Key Replacement Kia Sportage Key Replacement Replacement Kia Sportage Key
11 Ways To Completely Sabotage Your Anxiety Treatment
For Dogs anxiety treatment without medication
5 Must-Know Program Keys For Cars Practices For 2023
smart key programming near me
Why Car Boot Lock Repair Still Matters In 2023 Central Locking Repair Near Me
How To Save Money On Teaming Up With Birth Injury Attorneys birth injury legal professional (Lincoln)
5 Characteristics To Identify When Evaluating A Happy Hour
중구오피
Sofas Sectional Tools To Improve Your Daily Life Sofas Sectional
Technique Every Person Needs To Be Able To Sofas sectional
20 Trailblazers Setting The Standard In 3 Tier Bunk Bed
Triple Bunkbed
What’s The Current Job Market For Titration ADHD Medications Professionals?
titration Adhd medications
Why Locked Keys In Car Service Is More Difficult Than You Imagine Vehicle Unlocking
Five Killer Quora Answers On Washington Birth Injury
Attorneys Washington Birth Injury Attorneys
Are You Responsible For An Single Bed Bunk Beds Budget? 12 Tips On How To Spend Your Money Metal bunk bed single
Don’t Buy Into These “Trends” About Sectional Sofa L Shaped leather sofa l
shape (silichem.co.kr)
Sectional Sofa The Process Isn’t As Hard As You Think 4452346.xyz
The Fun Of Traveling And Tips On Buying Club Sets 대덕구오피
What Experts Say You Should Know car door lockout (Kattie)
20 Inspirational Quotes About Mesothelioma mesothelioma lawyer – Elvin,
9 Signs That You’re A Mesothelioma Attorneys Expert mesothelioma settlement
The Leading Reasons Why People Perform Well With The Wine Fridge Undercounter
Industry Wine fridge Built in, http://hoards.com.Cn,
14 Cartoons About Double Glazed Windows Near Me That Will Brighten Your Day
replacing
9 Things Your Parents Teach You About Designer Towel Radiators Designer towel Radiators
One Key Trick Everybody Should Know The One Vehicle Diagnostics Trick Every Person Should Know diagnostic Check
20 Things That Only The Most Devoted Adult Adhd Assessments Fans Understand adhd in adults assessment (Lashawnda)
Ten Ways To Build Your Treat Generalized Anxiety Disorder Empire anxiety treatment elderly
3 Wheel Mobility Trike: The Secret Life Of 3 Wheel Mobility Trike electric Three wheel scooter
What’s The Current Job Market For Best Detachable
Bunk Beds Professionals Like? best detachable bunk beds
What Will Glass Anal Butt Plugs Be Like In 100 Years? Small Anal Toy
Why Butt Plug Is The Right Choice For You? Sex buttplug
15 Of The Most Popular Pinterest Boards Of All Time About
Senseo Coffee Maker Senseo Coffee Machine
5 Reasons To Consider Being An Online Best Coffee Machine Pod Shop And 5 Reasons You Shouldn’t best rated coffee Machines
Collapsible Scooters: It’s Not As Difficult As You Think lightweight collapsible mobility scooters (p3terx.com)
20 Inspirational Quotes About Togel Singapore Togel Hongkong
9 Lessons Your Parents Taught You About Dripper Coffee Maker Dripper Coffee Maker
Incline Treadmill 101: The Ultimate Guide For Beginners electric Incline treadmill
A Look At The Future How Will The Test For Adult ADHD Industry Look Like In 10 Years?
How to Test adhd in adults
Fridge Freezers Built In Tools To Make Your Everyday Lifethe Only Fridge
Freezers Built In Trick Every Individual Should Know Fridge Freezers Built In (http://Www.Asystechnik.Com/Index.Php/Benutzer:EzraNapper0)
What’s The Current Job Market For Bandar Toto Professionals?
bandar toto
The 12 Best Robot Vacuum Mop Accounts To Follow On Twitter robot mop And Vacuum combo
10 Easy Steps To Start Your Own Bagless Self Emptying Robot Vacuum Business bagless self-emptying vacuums
Ten Things Your Competitors Help You Learn About Electric Fire Suite Cheap Electric Fire Suite
10 Quick Tips About Foldable Pram folding pram
10 Facts About Window Cambridge That Will Instantly
Put You In An Optimistic Mood plastic window repair
The Mesothelioma Attorney Success Story You’ll Never Imagine Mesothelioma Law
10 Things We All Love About Citroen C3 Key Fob Replacement replacement key For citroen c1
Your Family Will Thank You For Getting This Car Locksmith Near Me lock smith for car
15 Gifts For The Locksmith For Cars Lover In Your
Life mobile Locksmith car (wolvesbaneuo.com)
See What ADHD Adults Test Tricks The Celebs Are Using Adhd Adults Test
Is Your Company Responsible For The Key Reprogramming Budget?
Twelve Top Ways To Spend Your Money car key programming cost uk
Where Can You Find The Best Key Programming Near Me Information? car keys cut and programmed
4 Dirty Little Secrets About The Replacement Keys Car Industry Locksmith Car key replacement
What’s The Job Market For Mazda 6 Key Fob
Professionals? Mazda 2 Car Key Replacement
10 Healthy Fridges & Freezers Habits Fridge deals uk
How Contemporary Multi Fuel Stoves Has Changed The History Of Contemporary Multi
Fuel Stoves contemporary multi fuel Stoves 5kw
This Is The New Big Thing In Window Hinge Repair Near Me Replacement window hinges near me
See What Single Pushchair Sale Tricks The Celebs Are Using
single pushchair sale; http://dancelover.tv/node/27981,
Rollator Folding Tools To Streamline Your Daily Lifethe One Rollator Folding Trick
Every Individual Should Be Able To rollator folding
Loft Beds For Teens Tools To Ease Your Daily Life Loft Beds For Teens
Trick That Every Person Must Be Able To loft beds for teens (mail.Swgtf.com)
15 Things You’re Not Sure Of About Togel Resmi Indonesia Situs Toto Togel
5 Reasons To Be An Online Ford Car Keys Buyer And 5 Reasons
Not To repaired
10 Things Everyone Makes Up Concerning Small Wood Burner small Wood Burning stoves
A How-To Guide For Assessments For Adhd In Adults
From Start To Finish adhd evaluation
You’ll Never Guess This Fridge Freezer Sale’s Secrets Fridge Freezer sale
How Long Does 1kg Of Coffee Beans Last Techniques To Simplify Your Daily Life
How Long Does 1kg Of Coffee Beans Last Trick That Should Be Used By Everyone Know 1Kg Of Coffee Beans
14 Savvy Ways To Spend On Leftover Disabled Scooters Budget cheap Electric mobility scooter
20 Trailblazers Setting The Standard In Link Togel Toto Macau
See What Situstoto Slot Tricks The Celebs Are Using Situstoto slot
What’s The Current Job Market For Oil Filled Radiators Black
Professionals Like? Oil filled radiators Black
Three Greatest Moments In Childrens Bunk Beds With Stairs History 849827
Fireplace Bioethanol Tools To Improve Your Daily Lifethe One Fireplace
Bioethanol Trick That Should Be Used By Everyone Be Able To Fireplace Bioethanol
The Most Significant Issue With Twin Stroller, And How You Can Resolve
It twin Stroller side by side
What’s The Job Market For What Is The Best Automatic Folding Mobility
Scooter Professionals? What Is The Best Automatic Folding Mobility Scooter
Mesothelioma lawyers can assist victims in Oklahoma and their families receive
compensation for medical expenses as well as funeral expenses, lost wages and pain and suffering.
Feel free to visit my web site; asbestos lawyer
The Top Companies Not To Be Keep An Eye On In The New Audi
Key Industry audi car key holder – https://securityholes.science/wiki/Audi_A3_Keys_History_History_Of_Audi_A3_Key,
The 10 Most Terrifying Things About Private Psychiatrist Appointment Private Psychiatrist Appointment
The 9 Things Your Parents Teach You About Car Key Programmer key programmer (https://www.Question-ksa.com/user/susanstream65)
Five Killer Quora Answers To Situs Terpercaya Situs Terpercaya
5 Killer Quora Answers To Folding Treadmills folding treadmill
5 Killer Quora Answers To Kia Sportage Replacement Key kia sportage replacement key – Shay,
10 Double Glazing Repairs Near Me Meetups You Should Attend double glazed windows repair
What’s The Job Market For Deep Sectional Sofa Professionals Like?
deep Sectional sofa
How To Build A Successful ADHD Test For Adults When You’re Not Business-Savvy
female Adhd test
A Provocative Rant About Double Double Bunk
Beds Double Bunk bed with desk underneath
10 Facts About Holistic Treatment For Anxiety That Will Instantly
Get You Into A Great Mood stress and anxiety Treatment
Learn About Adult Adhd Assessment Near Me While You Work From At Home adhd Assessment for adults cost
Tips Regarding How To Get A Wingwoman 남구오피
The 9 Things Your Parents Teach You About Situs Togel Terpercaya situs Togel terpercaya
10 Reasons Why People Hate ADHD In Adults Test ADHD In Adults Test adults with adhd test (https://scientific-programs.science)
9 . What Your Parents Taught You About Indoor Ethanol Fireplace indoor ethanol Fireplace
What Is Private ADHD Titration And Why You
Should Take A Look what Is titration in adhd
Five Killer Quora Answers On Composite Door Panel Replacement Composite Door Panel Replacement
20 Myths About Window Glass Replacement: Debunked window double Pane glass replacement
The Secret Secrets Of Oil Filled Radiator Oil Portable oil filled radiator
The Leading Reasons Why People Achieve In The Treating Adult ADHD Industry symptoms of adhd in adults and treatment (Jane)
How To Survive Your Boss On Key Programmers car key programmers near me
You’ll Never Guess This Situstoto Slot’s Tricks Situstoto Slot
10 Sites To Help You Become An Expert In Repairs To Upvc Windows upvc window repairs (http://www.stes.tyc.edu.tw/xoops/modules/profile/userinfo.php?uid=1993133)
Where Will Togel4d One Year From Now? Situs togel online
12 Facts About ADHD Titration Private To Make
You Think About The Other People Private adhd medication titration
How To Tell If You’re Prepared To Go After Freestanding Fires floor standing electric fireplace
What’s Holding Back The Electric Treadmill Running Machine Industry?
best portable electric treadmill
Why Nobody Cares About Train Injury Lawsuit Train Injury lawyer
3 Ways The Adult Adhd Assessment Uk Can Influence Your
Life book adhd assessment uk (Terese)
The Best Way To Explain Double Glazed Near Me To Your Boss double glazed Window Units
10 Websites To Help You Be A Pro In Shark Robotic Vacuum Cleaner best robot vacuum and mop for luxury vinyl plank floors
3 Ways In Which The Window Repairs Cambridge Can Influence Your Life Upvc Door Repair Cost
Where Can You Get The Most Reliable Private Consultant Psychiatrist Information? private psychiatric Assessment Uk
10 Healthy Habits For A Healthy Toto Online Terbaik toto Macau
Tips For Explaining Ethanol Fireplace To Your Mom Bio ethanol fire
What Is Milton Keynes Double Glazing’s History?
History Of Milton Keynes Double Glazing replacement doors milton keynes
Question: How Much Do You Know About Womens Vibrating Panties?
panties that vibrate – https://pittman-Dwyer.blogbright.net/
–
20 Tips To Help You Be Better At 4 Wheeled Scooters 4 wheeler mobility scooter – Arlen
–
Ten Tree House Bunk Beds That Really Make Your Life Better Tree house bunk beds
20 Resources That Will Make You Better At Togel4d Togel Hongkong
The Sage Advice On Milton Keynes Door Panels From The Age Of Five commercial Units Milton keynes
10 Things You Learned From Kindergarden Which Will Aid You In Obtaining Citroen Key Replacement Near Me citroen Picasso Key fob programming
9 . What Your Parents Taught You About Togel4d Login togel4D
You’ll Never Be Able To Figure Out This 8mph Folding
Mobility Scooter Uk’s Tricks 8mph Folding Mobility scooter uk
10 Things That Your Family Teach You About Key Cutting Car
key Cutting car
10 Things We Love About Folding Wheelchair Ramp
wheelchair ramps vans
10 Life Lessons That We Can Learn From Greenpower Mobility Scooters Mobility Power Scooters
Small Bunk Bed For Kids Tools To Ease Your Everyday
Lifethe Only Small Bunk Bed For Kids Technique Every Person Needs To Learn Bunk Bed
Incontestable Evidence That You Need Automotive Locksmith Key Programming Automotive key Programmer
Private ADHD Assessment Explained In Less Than 140 Characters private adhd assessment Exeter
What’s The Job Market For French Door Fridge With Ice Maker Professionals Like?
best french Door fridge with ice and water Dispenser
The Intermediate Guide On Locksmith For Cars Locksmith for Cars near me
9 Signs That You’re An Expert All Terrain Buggy Uk Expert best all terrain Pram uk
Everything You Need To Learn About Automatic Locksmith automotive locksmith near Me prices – https://offmarketbusinessforsale.com/youll-Never-be-able-to-figure-out-this-automotive-locksmith-key-programmings-tricks/,
Why Is It So Useful? During COVID-19 ford key fobs; https://www.diggerslist.com/65e852821d13a/About,
Ten Travel Mobility Scooters-Related Stumbling Blocks You Shouldn’t Post On Twitter Motorized Scooters
See What Easy-To-Use Mobility Scooters Tricks The Celebs Are
Making Use Of Easy-To-Use Mobility Scooters (http://Spectr-Sb116.Ru/)
Do Not Buy Into These “Trends” About Treadmill Electric small electric treadmill for apartment (Astrid)
The 10 Most Terrifying Things About How Do I Get A Replacement Key For My Mitsubishi How do i get a replacement Key for My mitsubishi
The 10 Most Scariest Things About Situs Toto Login situs Toto Login
Guide To Volkswagen Replacement Key Cost Uk: The Intermediate Guide
Towards Volkswagen Replacement Key Cost Uk volkswagen replacement Key cost uk (dokuwiki.stream)
Nine Things That Your Parent Taught You About Veleco Scooter veleco
This Is What Free Standing Bioethanol Fires Will Look Like In 10
Years Time Freestanding fireplaces
Link Togel Techniques To Simplify Your Daily
Lifethe One Link Togel Trick That Should Be Used By Everyone
Be Able To Link Togel
5 Killer Quora Answers To Power Wheelchair Foldable Power Wheelchair Foldable
Five Pavement Mobility Scooters Uk Lessons Learned From Professionals pavement scooter
Guide To Situs 4d: The Intermediate Guide Towards Situs 4d
Situs 4d
The 9 Things Your Parents Teach You About Capsule
Coffee Machine Uk capsule coffee machine (Jane)
9 Lessons Your Parents Teach You About Rollator Walker Folding rollator walker folding (Passneurosurgery.net)
20 Reasons To Believe Situstoto Slot Will Never Be Forgotten Togel Hongkong
Folding Electric Lightweight Wheelchair: The History Of Folding Electric Lightweight Wheelchair In 10 Milestones attendant controlled folding electric wheelchair
What’s The Current Job Market For Automatic Fold Up Scooter Professionals?
Automatic fold up scooter
12 Companies Leading The Way In Coffee Machine Capsule capsule Coffee Maker
20 Private ADHD Assessments Websites Taking The Internet By Storm private Psychiatrist adhd assessment
What Pavement Mobility Scooters Uk Will Be Your Next Big
Obsession Scooter On Pavement
The Reason Behind Comfortable Mobility Scooters Is Everyone’s Obsession In 2023
Lightweight foldable scooters
Five Killer Quora Answers On Link Togel Resmi
link togel (Antoine)
It’s The One Fiat 500 Key Replacement Cost Trick Every Person Should
Be Able To Fiat key Duplication
How To Know If You’re Prepared For Folding Mobility Scooters For Sale foldable mobility scooter For adults
15 Reasons To Not Ignore Reprogramming Car Key Reprogramming Car Keys – Imoodle.Win,
15 Gifts For The Mobile Locksmith Auto Lover In Your Life auto locksmith key Programming near me
Don’t Be Enticed By These “Trends” About Single Mattress Topper Mattress pillow topper
5 Qualities People Are Looking For In Every Fiat Doblo Key Fob Replacement Fiat
500 Key Fob (Heavenarticle.Com)
5 Killer Quora Answers To Auto Folding Mobility Scooter With Suspension auto folding mobility scooter With Suspension
See What Lightweight Mobility Scooters For Sale Tricks The Celebs Are
Using Lightweight Mobility Scooters For Sale
15 . Things That Your Boss Wants You To Know
About Single Mattress Deals You Knew About Single Mattress
Deals single bed mattress cheap, Michel,
Five Killer Quora Answers On Best Class 3 Mobility Scooter Uk best class 3 mobility scooter uk [https://willysforsale.com/author/yogurtcover94/]
5 Killer Quora Answers On Situs Terpercaya situs Terpercaya
How Do I Explain Adhd Assessments For Adults To A Five-Year-Old Adhd assessment for women
U Shaped Sofa Explained In Less Than 140 Characters U shaped double chaise sectional
What Is Best Double Dildo And Why Is Everyone Talking About It?
Man and Woman double ended dildo
Bean To Coffee Machine: What No One Is Talking About Bean To Coffee Machines
Your Family Will Be Thankful For Having This Private Adult ADHD
Diagnosis Private Adhd Assessment Brighton Cost
It’s The Complete Guide To ADHD Assessment Private private adhd
assessment reading [80adec2Ampndbs9H.рф]
King Size Memory Foam Mattress Pad Tools To Ease Your Daily
Lifethe One King Size Memory Foam Mattress Pad
Trick That Everyone Should Know king size memory foam
mattress Pad (https://worldaid.eu.org/discussion/Profile.php?id=61616)
Nine Things That Your Parent Teach You About Machine Espresso Machine espresso
How To Become A Prosperous 5kw Wood Burning Stove Entrepreneur Even If You’re Not Business-Savvy stovesonline
Skoda Replacement Key Cost: What’s No One Is Discussing skoda octavia key Not working
The 10 Most Terrifying Things About How To Get A Replacement Honda Car Key
how to get a Replacement honda car Key
Situs Toto Tools To Make Your Everyday Lifethe Only Situs Toto
Technique Every Person Needs To Be Able To situs toto
(Olderworkers.com.au)
10 Quick Tips For SEO Consultancy London London seo firm, Rdvs.workmaster.ch,
Where Can You Get The Most Reliable Replacement Land Rover
Key Information? land rover Activity key review
What Is Coffee Grinder And Why You Should Be Concerned Ceramic Grinder
So You’ve Bought Electric Fireplace Heater …
Now What? electric log burner cream (glamorouslengths.com)
5 Laws That Will Help The Car Key Repair Service Industry Car key repair company (ai-db.science)
10 Things Everybody Hates About Freestanding Electric Fireplace electric fires free standing
11 Ways To Completely Sabotage Your Toto Result 안전놀이터 (Humanlove.stream)
Five Killer Quora Answers To Pragmatic Slots Free Trial 프라그마틱 불법
The Reasons You Should Experience Adult Adhd
Assessments At Least Once In Your Lifetime adhd assessment for adults Edinburgh (makeupstamp67.werite.Net)
You’ll Never Be Able To Figure Out This Oil Filled Radiator Oil’s Tricks Oil Filled Radiator Oil
The Top Reasons Why People Succeed In The Freestanding Fridge Industry fridge Freezer Uk
See What Window Repair Bromley Tricks The Celebs Are Using Window Repair Bromley
The Leading Reasons Why People Perform Well Within The Coffee
Machine Bean To Cup Industry best bean to cup Coffee Machine uk
Why Fireplace Electric Suite Is More Dangerous Than You Believed flat wall electric Fireplace suites
Mesothelioma is a rare cancer that occurs in the protective linings of certain organs.
It is most often found in the lung (pleura) but it can also develop in the chest wall or abdomen, or even in the testes.
My web site Asbestos Legal
10 Unexpected Capsule Coffee Maker Tips capsule coffee makers
The 9 Things Your Parents Taught You About Situs Togel Terpercaya Situs Togel terpercaya (http://Www.topsorb.com/)
A mesothelioma lawsuit or trust fund claim could
help Asbestos Attorney victims pay medical bills and other expenses.
Top law firms have attorneys who combine legal expertise and compassion.
Why Best Sex Toy For Clit Is Right For You?
clit Toys
You’ll Be Unable To Guess GSA SER Coupon’s Tricks gsa ser
coupon [Hollie]
15 Inspiring Facts About Ford Car Key Replacement The Words You’ve Never Learned ford key programming tool
7 Things About GSA SER Search Engine Ranker You’ll Kick Yourself For Not Knowing review
15 Gifts For Your Double Glazing Doctor Lover In Your Life skylight window Repair
Guide To Situs 4d: The Intermediate Guide For Situs 4d Situs 4D
Why Adding A Class 3 Mobility Scooters To Your Life’s Routine
Will Make The Different are electric mobility scooters allowed on the road
14 Businesses Doing A Great Job At Repairing Glass window glass repair service (Augustus)
You’ll Never Guess This Mobility Scooter Veleco’s Benefits Mobility scooter veleco
Should You Employ A Bad Credit Personal Home Finance Loan? 200만원 대출
What’s The Current Job Market For Renault Clio Key Card Replacement Professionals
Like? renault clio key card replacement; Dieter,
What NOT To Do When It Comes To The Pragmatic Slot Recommendations Industry 프라그마틱 플레이
The 10 Scariest Things About Replacement Key For Mini Cooper replacement Key For Mini
Five Killer Quora Answers To Remote Automatic Folding Scooter
Remote Automatic Folding Scooter
The Hidden Secrets Of Toto Korea Prize 스포츠토토 사이트
10 Things Everybody Has To Say About Bean To Coffee Machine bean-to-cup coffee machines
20 Myths About Pragmatic Slot Recommendations: Busted 프라그마틱 무료슬롯
The Top Reasons Why People Succeed In The Sports Toto Past Results Industry
토토사이트 (http://Www.Metooo.Io)
How The 10 Worst Modular Sleeper Sofa FAILS Of All Time Could Have Been Prevented sleeper sofas
10 Erroneous Answers To Common Wood Burners Questions:
Do You Know The Correct Ones? Woodburner outdoor
The 10 Most Terrifying Things About Fireplace Wall
Mount fireplace wall (tinted-lemon-Hfbm9G.mystrikingly.com)
5 Killer Quora Answers On Daftar Situs Togel Daftar Situs Togel
7 Helpful Tips To Make The Most Of Your Small American Fridge Freezer american fridge freezer sales uk
Where Will Pragmatic 1 Year From Right Now? 프라그마틱 환수율
The 10 Most Terrifying Things About Arabica Coffee Beans
1kg arabica coffee beans 1kg (Anne)
Don’t Stop! 15 Things About Freelander 2 Replacement Key We’re
Sick Of Hearing Land Rover Activity Key Price (https://Olderworkers.Com.Au/)
Guide To Lightweight Foldable Wheelchairs: The
Intermediate Guide For Lightweight Foldable
Wheelchairs lightweight Foldable wheelchairs
10 Facts About Electric Treadmill Cheap That Can Instantly Put You In An Optimistic Mood treadmill without electric
A Comprehensive Guide To Mesothelioma Law
Firm From Beginning To End Mesothelioma Lawsuit – Yogicentral.Science,
Do You Know How To Explain Mesothelioma Settlement
To Your Mom Mesothelioma Case (https://Dokuwiki.Stream/Wiki/This_Weeks_Top_Stories_About_Mesothelioma_Attorney)
How Small Single Mattress Is A Secret Life Secret Life
Of Small Single Mattress bed single mattress (Tristan)
Ten Stereotypes About Women’s Adult Toys That Don’t Always Hold Adult girls toys (https://www.medexmd.com/community/profile/lauri8599381560)
You’ll Never Guess This Single Buggy With Buggy Board’s Tricks single buggy with buggy board (Patricia)
“Ask Me Anything,” 10 Answers To Your Questions About Double Bunk Bed For Adults bunk double
bed with desk; Alma,
The 10 Most Scariest Things About Lost My Keys lost my Keys
What Private Psychiatrist Nottingham Experts Would Like You To Be Educated Private
psychiatrist wheathampstead (Short-korsgaard.hubstack.net)
10 Things Everyone Hates About Sports Toto 토토사이트 모음
12 Companies Are Leading The Way In Folding Mobility Scooters Near Me electric Mobility scooter foldable
Guide To Wine Cooler And Fridge: The Intermediate Guide
To Wine Cooler And Fridge wine cooler And Fridge
What NOT To Do In The Sectional Sofas Industry green sectional sofa
20 Trailblazers Lead The Way In Lightweight 3 Wheel Rollator With Seat Senior Mobility Aid
20 Tools That Will Make You More Effective At Veleco
Luxury Electric Scooters veleco electric scooter
Mesothelioma, a deadly cancer, is largely caused by exposure to asbestos.
A New York mesothelioma lawyer can assist victims and their families in holding companies accountable for concealing Asbestos Lawyer, a
dangerous chemical.
14 Cartoons On Mid Century Leather Sofa Which Will Brighten Your Day pure Leather sofa
3 Ways The Bluetooth Sex Machine Can Affect Your Life Sexual Machine
How Do You Explain Lovense Fucking Machine To A Five-Year-Old bondage Machines
What Is Nissan Juke Spare Key And Why Is Everyone Speakin’ About It?
nissan pulsar key Replacement
You’ll Never Guess This Adults ADHD Test’s Secrets Adults Adhd Test
This Is The Ugly Reality About Lost Lexus Key Fob 2001 Lexus ls430 Key replacement
20 Important Questions To ASK ABOUT Fridge Uk Before You Buy Fridge Uk buy fridge uk
How 3 Wheel Lightweight Mobility Scooters Changed Over Time Evolution Of 3 Wheel Lightweight
Mobility Scooters 3 wheeled electric scooters (Lesli)
Think You’re The Perfect Candidate For Doing L Shape Bunks?
Take This Quiz l-shaped bunk Beds
Are You Responsible For An Couches For Sale Budget?
12 Ways To Spend Your Money black couches for Sale
10 Tips For Machines Espresso That Are Unexpected professional espresso machine for home (Willa)
The Biggest “Myths” About Porsche Macan Key Could Actually Be True Porsche car keys made
25 Unexpected Facts About Toto Macau situs toto (http://www.Google.com.ag)
10 Things That Your Family Teach You About Veleco Mobility Scooter Reviews veleco mobility scooter reviews
A Sage Piece Of Advice On ADHD Women Test From An Older Five-Year-Old add Women
Guide To Daftar Akun Togel Resmi: The Intermediate Guide In Daftar Akun Togel Resmi Daftar akun togel Resmi – http://www.wulanbatuoguojitongcheng.com,
Its History Of Nissan Key Programming How To Get A New Nissan Key
A Proactive Rant About Volvo Replacement Key volvo v40 key programming (http://dudoser.Com)
Pragmatic Slot Manipulation Tips From The Top In The Business 프라그마틱 슬롯 체험
The Most Hilarious Complaints We’ve Seen About G Spot Sex Toy G spot sex toys (https://lovewiki.faith)
5 Killer Quora Answers To Bunk Beds Kids bunk beds
kids (https://bookmarkfeeds.stream)
The 10 Scariest Things About Cheap Sofas For Sale Cheap sofas for sale
The Little-Known Benefits Of Pragmatic Slots Return Rate 슬롯
Guide To Coffee Machines For Nespresso Pods: The Intermediate Guide In Coffee Machines For Nespresso Pods Coffee machines for
nespresso pods (Escortexxx.ca)
Guide To GSA SER Services: The Intermediate Guide In GSA SER Services gsa ser services, Alysa,
20 Rising Stars To Watch In The Sectional Sofa Sale
Industry Sectional sale
See What Ferrari Key Replacement Uk Tricks The Celebs Are Utilizing ferrari key replacement uk
A Brief History Of The Evolution Of Mazda Key replaced
A Time-Travelling Journey: How People Talked About Squirting Dildos 20 Years Ago Toys To Help You Squirt
Say “Yes” To These 5 Brown Squirting Dildo Tips http://Www.Topsadulttoys.Uk
Ten Fireplace-Related Stumbling Blocks You Shouldn’t Share
On Twitter electric fireplace (gitlab-sv.metechsoftware.Com)
See What Bulk Arabica Coffee Beans Tricks The
Celebs Are Utilizing bulk arabica coffee beans
14 Businesses Are Doing A Fantastic Job At Multi Fuel Stoves For Sale 34630194 (Francine)
How Bunk Bed For Children Has Become The Most Sought-After Trend In 2023 bunk beds usa
What’s The Most Common Cheap Rabbit Vibrators Debate Isn’t As Black And White As You Think
Adult rabbit toy
5 Common Myths About Assessment Of Adult Adhd You Should Avoid Adhd Assessment For Adults Cost
14 Smart Ways To Spend Left-Over American-Style Fridge Freezers Budget sale on fridge freezers uk (Francisca)
5 Laws Anyone Working In Live Casino Should Know 프라그마틱 슬롯버프 – http://47.108.249.16/home.php?mod=space&Uid=1677619 –
The 10 Most Scariest Things About Retro Fridge Freezer Sale
Retro Fridge Freezer Sale – https://Mcclain-Warner.Federatedjournals.Com/The-Best-Retro-Fridge-Freezer-Methods-To-Rewrite-Your-Life –
A Positive Rant Concerning Class 3 Mobility Scooter
class 3 mobility scooter uk [Eduardo]
Small Sleeper Sofa: What’s The Only Thing Nobody Is Discussing King Sleeper Sofa
10 50/50 Freezer Fridge That Are Unexpected 50/50 fridge – https://fridge-freezers56963.blogginaway.com/29911896/15-pinterest-boards-that-are-the-best-Of-all-time-about-50-50-fridge-freezer-integrated-frost-free –
Desk Treadmill Foldable Tools To Ease Your Daily Lifethe
One Desk Treadmill Foldable Trick That Should Be Used By Everyone Learn desk treadmill foldable (Lance)
Five Killer Quora Answers To Car Boot Scooters car boot scooter (Linnea)
Why Single Memory Foam Mattress Isn’t A Topic That People Are Interested In. bunk bed mattress [Lucinda]
A Handbook For West Elm Pull Out Couch From Start To Finish reclining sectional with pull Out bed [https://ghasemtorabi.ir/User/TammaraX59/]
The 10 Most Terrifying Things About Fold Up Wheelchairs fold up wheelchair
10 Things Everyone Hates About Pragmatic Play 프라그마틱 플레이
What Is Wall Mounted Fireplaces And Why Is Everyone Talking
About It? Electric wall mounted fireplaces
(socialwoot.com)
10 Things We Hate About Hoover Fridge Freezer With Water Dispenser
integrated fridge freezer With water dispenser
What’s The Job Market For Automatic Folding Lightweight
Mobility Scooter Professionals? automatic folding lightweight mobility Scooter
The 9 Things Your Parents Teach You About
Bed With Couch bed With Couch
Five Essential Qualities Customers Are Searching For In Every Kids Beds Bunk Beds
bunk bed for kids (Johnny)
This Week’s Top Stories Concerning Sectional Sofas sectional sofas with recliners (Ivey)
Check Out: How Hyundai I30 Key Replacement Is Taking Over And What Can We Do About
It how to Find lost hyundai key fob
50 50 Integrated Fridge Tips To Relax Your Daily Lifethe One 50
50 Integrated Fridge Technique Every Person Needs To Be Able To 50 50 integrated fridge [Nestor]
What Is Toto Online Terbaik And How To Utilize
What Is Toto Online Terbaik And How To Use Bandar Togel terpercaya
(exactlybookmarks.com)
Responsible For An Womens Panty Vibrators Budget?
Twelve Top Tips To Spend Your Money Sex with panty
15 . Things That Your Boss Wished You’d Known About Double Over
Double Bunk Bed double bed bunk beds with desk (Francesca)
How The 10 Worst Mens Sex Toys-Related FAILS Of All Time Could Have Been Prevented menssex toys
Five Killer Quora Answers On Composite Door Glass Replacement composite door glass replacement
The Best G Spot Vibrators Tricks To Transform Your Life best G spot vibrators (ottawa.pinklink.ca)
What’s The Current Job Market For 3 Wheel Rollator Walker With
Seat Professionals? 3 wheel rollator walker with seat [Jada]
The Best Way To Explain Nescafe Classic 1kg To Your Mom coffee 1kg (Faustino)
20 Trailblazers Lead The Way In Squirting Dildo Toy topsadulttoys
Why Nobody Cares About Audi A3 Key Battery Audikey
8 Tips To Improve Your Stylish Mobility Scooters Game lightweight foldable
scooters; Kirby,
What You Can Use A Weekly Best Coffee Machines Project Can Change Your Life best nespresso
Coffee machine [dudoser.com]
Technology Is Making Ghost Immobiliser Review Better Or Worse?
immobilisers
Replacement Lock For Composite Door Tools To Streamline Your Daily Lifethe One Replacement Lock For Composite Door Trick That Every Person Must
Know replacement lock for composite door
Five Killer Quora Answers To Treadmills UK treadmills uk (Cynthia)
14 Common Misconceptions About Modern Leather Sofa contemporary leather sofa (Kathy)
10 Unexpected Bed Double Mattress Tips double bed mattresses
Say “Yes” To These 5 Vauxhall Car Key Tips new vauxhall Car key
Who Is Responsible For An Electric Foldable Treadmill Budget?
12 Tips On How To Spend Your Money folding electric
treadmill (Maricela)
What The 10 Most Worst Realistic Sex Doll For Sale Mistakes Of All
Time Could Have Been Prevented real Life sexdoll
Guide To Samsung American Fridge Freezer With
Water And Ice Dispenser: The Intermediate Guide The Steps To
Samsung American Fridge Freezer With Water And Ice Dispenser Fridge Freezer With Water And Ice (Glamorouslengths.Com)
What’s The Reason Porsche Key Shell Is Fast Becoming The Hottest Fashion Of 2023 Porsche Cayenne Keys
Locked In Car (Telegra.Ph)
3 Reasons Commonly Cited For Why Your Free
Slot Pragmatic Isn’t Performing (And Solutions To Resolve It) 프라그마틱 슬롯 환수율 (Avery)
20 Things That Only The Most Devoted Double Glazed Replacement Glass Near Me Fans Know double glazing near me
(http://spectr-sb116.ru)
Five Killer Quora Answers To Fridge Freezer 50 50 Sale fridge
freezer 50 50 sale – Nicholas,
The Reasons Vibrating Panties Isn’t As Easy As You Imagine Vibrator panties
From All Over The Web: 20 Fabulous Infographics About Cheap Squirting Dildo
Topsadulttoys.Uk
You’ll Never Be Able To Figure Out This Wild Harvested Arabica Coffee
Beans’s Secrets Wild Harvested Arabica Coffee Beans
10 Tips For Quickly Getting Sports Toto History 토토사이트
How To Know If You’re Set To Go After Drip Filter Coffee
coffee maker drip (Porfirio)
What Is The Reason? Frost Free Fridge Freezer 50/50 Sale Is Fast Increasing To Be The Trendiest Thing Of 2023 upright fridge freezer 50 50; glasstool.kr,
Coffee Beans Coffee Machine: 11 Things You’re Not Doing best automatic bean to cup coffee machine (Reynaldo)
The Three Greatest Moments In Coffeee Machine History coffee machines best (worldaid.eu.org)
Guide To Fridge Freezer Frost Free 50/50: The Intermediate Guide Towards Fridge Freezer Frost Free 50/50 fridge freezer frost
free 50/50 (Johnie)
3 Reasons 3 Reasons Why Your Filter Coffee Is Broken (And
How To Fix It) Filter & Drip Coffee
Guide To 3 Wheel Rollator With Seat Uk: The Intermediate Guide In 3 Wheel Rollator With Seat Uk 3
wheel rollator with seat uk (Mikki)
Five Killer Quora Answers On Affordable Link Building Services Affordable link Building services
How To Explain Seat Leon Car Key To A Five-Year-Old
seat leon key programming
What’s The Current Job Market For Composite Door Paint Repair Professionals Like?
composite door paint repair
What’s The Current Job Market For Upvc Window Handle
Replacement Professionals? window handle Replacement
10 Beautiful Images Of Land Rover Key land rover discovery sport key not working
(Leon)
15 Reasons To Not Ignore Seat Leon Key Fob Replacement seat mii replacement key
– John,
How To Become A Prosperous Attorney Asbestos If You’re Not Business-Savvy asbestos lawyers
– dlightcompany.co.Kr,
What Adult Adhd Symptoms Women Experts Want You To Learn adult adhd symptoms men – Callum,
Online Dating Tips Rookies 오피사이트 [http://253308.cn/home.php?mod=space&uid=5223326]
Why No One Cares About Asbestos Attorney asbestos attorneys (Hallie)
Five Window Replacement Cost Lessons From The Professionals window replacement Cost uk (wayranks.com)
See What Asbestos Attorneys Tricks The Celebs Are Utilizing asbestos attorneys (Jorge)
“Ask Me Anything:10 Responses To Your Questions About Asbestos Mesothelioma mesothelioma attorneys (Kristin)
Why We Our Love For Washing Machine 10kg Uk (And You Should Too!)
023456789
5 Killer Quora Answers To Replacement Double Glazing Units Near Me
double glazing units near me
What’s The Current Job Market For Electric Wheelchair Heavy Duty Professionals Like?
electric wheelchair heavy duty (cheap-mobility-scooters45554.blogkoo.com)
The 10 Most Dismal Private ADHD Errors Of All Time Could Have Been Prevented Private ADHD assessment Hampshire
Why You Should Focus On Improving Wall Mounted Electric Fires wall Hung electric fireplace
The 10 Most Scariest Things About Link Togel link togel (Sophie)
13 Things About Asbestos Attorney Mesothelioma You May Not Have Known mesothelioma lawsuits, Tangela,
5 Killer Quora Answers On Composite Door Frame Replacement
Composite door frame replacement
You’ll Never Be Able To Figure Out This Togel Hongkong’s Tricks
togel Hongkong
Guide To Door Doctor: The Intermediate Guide On Door Doctor Door Doctor
What Is The Reason Pragmatic Slots Site Is The Right Choice For You?
프라그마틱 체험 (King)
Are You Responsible For The Pragmatic Korea Budget?
10 Ways To Waste Your Money 프라그마틱 무료체험 슬롯버프 (Rashad)
The Most Advanced Guide To Mesothelioma Asbestos Mesothelioma attorney (http://www.jsgml.top)
Poor Credit Personal Loans Up To $1500 In 24 Hours 신혼부부 대출
5 Vauxhall Key Fob Lessons Learned From The
Professionals vauxhall Keys cut
10 Top Facebook Pages Of All-Time About Ultralight Electric Wheelchair electric wheelchair chair
(Christen)
11 Strategies To Completely Block Your Private Psychiatrist
Belfast how much does it cost to see a private psychiatrist (Cleta)
Are You In Search Of Inspiration? Try Looking Up Car Diagnostic Near Me car diagnostic
tests – Nannie,
10 Rabbit Vibrator For Sale Tips All Experts Recommend Rabbit vibrator toy
The 9 Things Your Parents Taught You About Window Hinge Repairs Near Me window hinge repairs near me (https://championsleage.Review/)
20 Reasons To Believe Private Psychiatrist Sheffield Cost Will Never Be Forgotten private psychiatry northern ireland (Lashay)
What Is Cheapest Realistic Sex Doll And How To Utilize What Is
Cheapest Realistic Sex Doll And How To Use realistic sex dolls review (morgh-online.ir)
Double Glazing Door Handles: 10 Things I’d Like To Have Known Sooner upvc chrome door handles (Micheal)
The Best Automatic Folding Mobility Scooters Tricks To Make A Difference In Your Life best automatic folding mobility
scooter (Dong)
What’s The Reason Everyone Is Talking About How Much Is A Private
ADHD Assessment UK Right Now private adhd assessment oxford
(Victorina)
How To Become A Prosperous Bluetooth Fuck Machine Even If You’re
Not Business-Savvy male sex machines (telegra.ph)
Five Killer Quora Answers On Patio Door Locks Repair Patio Door Locks Repair
15 Terms Everybody Who Works In Situs Togel Dan Slot
Terpercaya Industry Should Know situs togel terpercaya (militarymuster.ca)
10 Healthy Habits To Use Kia Key Fob keys locked in kia optima (moon.gandme.co.kr)
9 Lessons Your Parents Teach You About Bunk Beds
For Children bunk (Orlando)
Replacement Volkswagen Key: Myths And Facts
Behind Replacement Volkswagen Key Volkswagen polo key replacement (telegra.ph)
15 Things You’re Not Sure Of About Car Diagnostics Near Me
workshop
See What Treadmills For Sale UK Tricks The Celebs Are Using Treadmills For Sale Uk
Private Mental Health Assessment: A Simple Definition Psychiatrist Mental Health Assessment (https://Cameradb.Review)
15 Seat Replacement Key Cost Benefits You Should All Be Able To seat ibiza key programming – Cathryn –
Seven Reasons Why Bentley Bentayga Key Is Important bentley key case, Lionel,
What’s The Current Job Market For Male Masturbating Toy Professionals Like?
male masturbating toy – Elsie,
A Help Guide To Double Glazing Windows Birmingham From Beginning To End french doors birmingham,
go-god.Main.Jp,
Double Glazing Repair Aylesbury It’s Not As Hard As You Think
Upvc door repairs near me
9 . What Your Parents Taught You About Local SEO Services Uk Local seo services Uk
The Benefits Of Mental Health Assessment At The Very Least
Once In Your Lifetime Mental Health Mood Assessment
See What Lightweight Folding Mobility Scooters Tricks The Celebs Are Making
Use Of Lightweight Folding Mobility Scooters
You’ll Never Guess This Situstoto Slot’s Tricks Situstoto slot
Upvc Repairs Near Me Tools To Ease Your Daily
Life Upvc Repairs Near Me Trick That Should Be Used By Everyone Learn Upvc Repairs near Me
20 Questions You Must Always Ask About Wheelchair Electric Wheelchair Before Buying
It Power chairs For handicapped (https://willysforsale.Com/author/fridaytown37/)
How Lightweight 3 Wheel Stroller Became The Hottest Trend Of 2023 3 wheeler pushchair off
road (Anja)
How To Build A Successful Replacement Upvc Door Panels Entrepreneur Even If You’re Not Business-Savvy
replacing upvc door locks (Joey)
10 Ways Casinos Fight Card Counters 보증금 대출 (wayranks.com)
What’s The Current Job Market For French Door Fridge With Ice Maker Professionals Like?
french door fridge with ice maker, Nicolas,
The History Of High Functioning Anxiety Disorder drugs For generalized anxiety disorder
Lost Key Replacement Car Techniques To Simplify Your Daily Lifethe One Lost Key Replacement Car Technique Every Person Needs To Learn lost key replacement car
Patio Door Track Repair Tools To Make Your Daily Lifethe One Patio Door Track Repair Trick Every Person Should Be Able To
patio door track Repair
5 Killer Quora Answers To Mobility Scooter To Buy Near Me Mobility Scooter To Buy Near Me
ADHD Test Adults: A Simple Definition online Adhd test
See What How Much Is A Private ADHD Assessment UK Tricks The Celebs Are Making Use Of how much is
a private adhd assessment uk [Roscoe]
15 Local Auto Locksmith Bloggers You Must Follow auto lock smith near me
10 Things You Learned In Kindergarden That Will Help You Get Local
Car Locksmith Locked Keys In Car Locksmith
You’ll Never Guess This Treadmills With Incline’s Benefits fit
Locksmith For Cars Isn’t As Difficult As You Think locksmiths for car keys near me – https://deleon-dehn-2.blogbright.net/20-reasons-why-locksmith-for-Car-will-never-be-forgotten,
See What Treadmills Best Tricks The Celebs Are Using
treadmills best [Millie]
These Are The Most Common Mistakes People Make With Key Programming car key programming (Iona)
15 Up-And-Coming Test ADHD In Adults Bloggers You Need To Watch adhd Testing uk
Patio Door Track Repair Tools To Improve Your Daily Life Patio Door Track Repair Trick That
Everyone Should Know patio door track repair
3 Ways The 3 Wheel Compact Stroller Influences Your Life 3 wheel running buggy
(Joycelyn)
Why We Why We Auto Locksmith (And You Should Also!) auto Locksmiths [https://auto-locksmiths06422.newbigblog.com/]
Integrated French Style Fridge Freezer Tips To Relax Your Daily Lifethe One
Integrated French Style Fridge Freezer Trick That Everyone Should Know integrated french style
fridge freezer (Anastasia)
Bandar Online Togel Tools To Streamline Your Everyday Lifethe Only Bandar Online Togel Trick That Every Person Must Learn bandar online togel
A Proficient Rant Concerning Pragmatic Product Authentication 프라그마틱 이미지
10 Things You Learned In Kindergarden That’ll Help You With Composite Door Hinges Adjustment Upvc window hinge repair
This Story Behind Spare Audi Key Will Haunt You For The Rest Of Your Life!
audi a1 key fob (Bettye)
What Can A Weekly Situs Terpercaya Project Can Change Your Life situs togel Online
Five Killer Quora Answers On Replacement Key For Audi A3 replacement key for audi, Beulah,
11 Creative Ways To Write About Wall Electric Fireplace wall hung electric fireplaces (Delilah)
The Private Psychiatrist Chester Awards: The Most Sexiest,
Worst, And Weirdest Things We’ve Seen Private psychiatrist cost [http://Www.stes.tyc.edu.tw]
See What Ghost Installer Tricks The Celebs Are Utilizing Ghost Installer
Best Childrens Bunk Beds Tools To Help You Manage Your
Daily Lifethe One Best Childrens Bunk Beds Trick Every Individual Should Be Able
To childrens bunk Beds
Why Pragmatic Slot Experience Is Fast Becoming The Most Popular Trend In 2024 프라그마틱 카지노
20 Resources To Make You More Effective At French Door Fridge
With Ice Dispenser french door fridge height (Henry)
A Glimpse Into Mini Cooper Car Key Replacement’s Secrets Of Mini Cooper Car Key Replacement Mini cooper key replacement near me
15 Up-And-Coming Replacement Key For Audi Bloggers You Need To Watch replacement key For audi a4
You’ll Never Guess This Automatic Folding Mobility Scooter Near Me’s Benefits automatic
folding mobility scooter near me (https://wu-woodward-2.hubstack.net)
Who Is Bunk Bed For Children And Why You Should Consider Bunk Bed For Children bunk beds uk
Wall Mounted Electric Fireplaces 10 Things I’d Like To Have Known Sooner wall fires electric (Quyen)
10 Meetups About ADHD In Adults Women You Should Attend Adhd In Adult Women Treatment
10 No-Fuss Ways To Figuring The Best Sex Toys For Men You’re Looking For best toys for men; Amparo,
15 Of The Most Popular Keys For Bmw Bloggers
You Should Follow Bmw Key 5 Series (Championsleage.Review)
Guide To Landrover Key Replacement: The Intermediate Guide The Steps To Landrover Key Replacement
landrover Key replacement
Why Electric Treadmill With Incline Will Be Your Next Big Obsession electric Treadmills
The Internet Site Business – A Quick Guide jusojula (https://writeablog.net/judoarcher2/the-trusted-ckan-and-knowledge-portal-skilled)
What’s The Current Job Market For Best Coffee Machine With
Milk Frother Professionals? best Coffee machine with Milk frother
(http://www.easyfie.com)
10 Things Your Competitors Teach You About
Replacement Key For Audi replacement audi A3 key
This Is A Saab Replacement Keys Success Story You’ll Never Be Able
To saab key programming near me (Lupita)
Replacement Audi Key Tools To Make Your Daily Lifethe One Replacement Audi Key
Trick That Every Person Must Learn Replacement Audi Key
The Next Big Thing In Toto 4d 토지노 커뮤니티
5 Assessment For Mental Health Projects For Any Budget
online mental Health Assessment
The 10 Most Terrifying Things About Automotive Locksmiths Near Me locksmith Automotive
A Look At The Future: What Will The Glass Replacement Industry Look Like In 10 Years?
Replacement Glass In Windows (https://Smith-Stout-3.Federatedjournals.Com)
Guide To Red Retro Fridge Freezer: The Intermediate Guide On Red Retro Fridge Freezer Retro Fridge Freezer
A Sage Piece Of Advice On Replacing Lost Car Keys From The Age Of Five remote
Five Qualities That People Search For In Every Ford Key Replacement Near Me ford keys replacement
11 “Faux Pas” You’re Actually Able To Create Using Your Professional SEO
Agency parasite
Be On The Lookout For: How Private Psychiatrist Cardiff Cost Is Taking Over And
What You Can Do About It Private Psychiatric Assessment Uk
You’ll Never Guess This Realistic Sexual Dolls’s Benefits sexdoll realistic
14 Cartoons On Realsexdoll Which Will Brighten Your Day realsexdolls
How To Make A Profitable Sleeper Sofa With Storage If You’re Not
Business-Savvy Customized Sleeper Sofas
The 10 Most Dismal Vehicle Door Lock Repair Mistakes
Of All Time Could Have Been Prevented Lock Fixing Near Me
Cabin Beds With Storage’s History History Of Cabin Beds With Storage
Cabin Beds Adults, Yerliakor.Com,
17 Reasons Not To Not Ignore Adhd Assessment Uk adhd assessment in Uk
(https://socialbaskets.com/)
10 Sites To Help You Develop Your Knowledge About Upvc Door Panels Replacement Upvc Door Panels Near Me
10 Things We All Are Hateful About Situs 4d toto macau (Xiomara)
The People Who Are Closest To Senseo Have Big Secrets To Share senseo coffee
10 Treadmills On Sale-Related Projects That Stretch Your Creativity Treadmills for Sale
15 Interesting Hobbies That Will Make You Smarter At Pragmatic Slots Free Trial
프라그마틱 무료슬롯
7 Simple Changes That’ll Make The Biggest Difference In Your Toto Korea 토토사이트 추천
10 Places That You Can Find 3 Wheel Compact Stroller 3 Wheel pushchair sale
What’s The Most Common Upvc Window Replacement Hinges Debate It’s Not As Black
And White As You Might Think patio door hinge replacement
You’ll Be Unable To Guess Sports Toto Special Draw’s
Benefits 토토사이트 순위 – Hermine,
5 Laws That Can Help In The Pragmatic Site Industry 프라그마틱 정품확인
A Look At The Future How Will The Asbestos Exposure Attorney Industry Look Like In 10 Years?
Asbestos Attorney – Funsilo.Date,
The Reason Why Fireplace Tools Set Is Everyone’s Passion In 2023 Electric Fireplace
The Ultimate Glossary Of Terms About Double Glazing Seal Repairs repairing Double Glazing
Why Link Togel Is Everywhere This Year link togel resmi (Estella)
How To Survive Your Boss In Audi Key Audi Replacement Keys
Could Assessment For Adhd In Adults Be The Key To
2023’s Resolving? get adhd Assessment (iampsychiatry74452.wikiadvocate.com)
Personal Loans- For Any And Every Personal Need 무직자 대출 쉬운곳
How Private Diagnosis ADHD Became The Hottest Trend In 2023 private Adhd assessment uk cost
The 10 Scariest Things About Upvc Patio Door Repairs Near
Me Upvc Patio Door Repairs Near Me
You’ve Forgotten Audi Key: 10 Reasons Why You Do Not Need It audi Key Replacement
5 Killer Quora Answers To Couches With Recliners couches with recliners
Playing Casino Poker For Your First Time 쥬라기월드 에볼루션 (mozillabd.science)
Why Mesothelioma Not Caused By Asbestos Might Be Your Next Big
Obsession mesothelioma Lawsuit
Wood Burner Stoves Uk: What’s New? No One Is
Talking About wood burning Stoves
Are You Responsible For A Adhd In Women Symptoms Budget?
12 Top Ways To Spend Your Money odd Adhd symptoms
Directory Submission – The Experience 링크모음, https://writeablog.net/,
10 Misconceptions Your Boss Has About ADHD Private Diagnosis
how to get diagnosed With adhd in adulthood
Why You Should Focus On The Improvement Of ADHD Treatment For Adults treatment for
adhd (https://mentalhealth62939.wikiusnews.com/885006/how_to_tell_if_you_re_set_to_go_after_treatment_for_adult_adhd)
10 Inspirational Graphics About Small Sleeper Sofa King sleeper sofa
What Is The Reason? Vehicle Diagnostics Is Fast Becoming The Hottest Trend Of 2022?
how much is car diagnostic test
Fireplace Surround: What No One Is Talking About Modern Fireplace (60.250.156.230)
Treadmills For Sale UK: What No One Is Talking About home fitness
10 Signs To Watch For To Buy A L Shaped Double Bunk Beds l shaped bunk bed with storage
10 Real Reasons People Hate ADHD Untreated In Adults Treatment
For Add Adhd In Adults (Psychiatry52115.Shivawiki.Com)
Why Lexus Key Is The Right Choice For You? Lexus key fob
The Secret Secrets Of Audi Spare Key Audi q5 key
Mesothelioma occurs when asbestos legal (Dee) fibers are
introduced into the body. The most well-known mesothelioma type is called
mesothelioma pleural, which develops in the chest cavity’s lining and lungs.
10 Mobile Apps That Are The Best For Sports Toto Near Me Today
토토사이트 순위
Five Killer Quora Answers To Electric Treadmill Vs Manual electric treadmill vs Manual
Bifold Door Replacement Tools To Ease Your Everyday Lifethe Only Bifold Door Replacement Trick Every Individual Should Be
Able To bifold door replacement
Guide To Treadmills Near Me: The Intermediate Guide
Towards Treadmills Near Me treadmills near me (Jermaine)
What Is The Reason Women Adult Toys Is The
Right Choice For You? best sex toys For girls
10 Life Lessons We Can Take From Togel4d Login Toto macau
Small Wine Chiller Tools To Make Your Everyday Lifethe Only Small Wine Chiller Trick That Every Person Should Know Small Wine chiller
Is Technology Making Toto Rules Better Or Worse? 메이저사이트 바록가기
A Look At The Myths And Facts Behind Mystery Box Mystery box Opening
Cat Flap Glass Door Installation Near Me cat flap glass door installation near me
Why Everyone Is Talking About How To Get Diagnosed With ADHD UK Right Now how to get adhd
diagnosis uk adults (Kevin)
You’ll Be Unable To Guess Pushchair Single’s Tricks pushchair single; Cortez,
Where Will Private Adult Adhd Assessment Be 1 Year
From Now? Adhd assessment score
The Most Common ADHD Diagnosis In Adults Mistake Every Beginning ADHD Diagnosis
In Adults User Makes Adhd diagnosis Questionnaire
This History Behind Second Hand Couches For Sale Will Haunt You Forever!
Sofa Sofa Sale, https://Wikimapia.Org/,
You’ll Be Unable To Guess Window Doctor Near Me’s Benefits near
This Week’s Most Remarkable Stories Concerning Double Glazing In Maidstone Garage door repairs maidstone
The 10 Most Terrifying Things About Locksmiths Cars locksmiths car (https://doodleordie.com/profile/dollbench32)
15 Top Pinterest Boards From All Time About Strollers 2 In 1
2 in 1 Stroller
20 Questions You Should Have To Ask About Free Standing Electric Fireplace Prior To Purchasing Free Standing Electric Fireplace wooden fireplaces
asbestos case (Rocky) attorneys can help families get compensation from companies that exposed them to
asbestos. They have experience in filing a variety of
lawsuits including personal injury and wrongful deaths claims.
10 Facts About Social Anxiety Symptoms That Will Instantly Get
You Into A Great Mood separation anxiety symptoms – Patricia
–
Why Do So Many People Want To Know About Retro Fridge Freezer
Cream? user-Friendly side by Side fridge freezer, howe-mccullough-2.mdwrite.net,
20 Great Tweets From All Time Replacement Double Glazing Units Near Me repair double glazed Window
Are Mobile Automobile Locksmith The Best Thing There Ever Was?
auto mobile locksmith near me – https://thejillist.com/,
You’ll Be Unable To Guess Double Glazing Windows Handles’s Benefits Double glazing windows handles
Guide To Fold Ramp For Wheelchair: The Intermediate
Guide In Fold Ramp For Wheelchair fold Ramp for Wheelchair
The Top Reasons Why People Succeed On The Landrover Key Replacement Industry Spare Land Rover Key
See What How Much Is A Private ADHD Assessment UK Tricks The Celebs
Are Making Use Of Private Adhd assessment Uk
See What Ferrari Key Fob Replacement Uk Tricks The Celebs Are
Utilizing Ferrari key fob
Take Control Over Your Environment – Dating Advice For Guys
강서오피 (anotepad.com)
9 Things Your Parents Teach You About Cheap Treadmill With Incline cheap treadmill with
incline (incomedegree0.bravejournal.net)
20 Things You Must Know About ADHD Private Diagnosis private adhd Assessment London Cost
10 Things That Your Family Taught You About Daftar Akun Togel Resmi
daftar akun togel resmi
Bull Bars On Four Wheel Drives 대구유흥 (writeablog.net)
Why Affordable SEO London Is So Helpful During COVID-19 seo London review
Do Not Make This Blunder On Your Double Glazing Windows Repairs double glazed windows Repairs
What Is The Reason Mazda 3 Key Is The Best Choice
For You? local
Guide To Replacement Key For Audi: The Intermediate Guide In Replacement Key For Audi
Key for audi
Guide To Replacement Key For Audi: The Intermediate Guide Towards Replacement Key For Audi Replacement Key For Audi –
Suzukiforum.Lv,
Looking For Inspiration? Look Up Locksmith Near Me Auto 24 hour auto locksmith near me
Cat Flap Double Glazing cat flap double glazing (dougherty-bernard-5.technetbloggers.de)
Responsible For A Wall Electric Fireplace Budget?
12 Best Ways To Spend Your Money fireplaces electric
20 Myths About Replacement Double Glazed Sealed Units: Dispelled Aluminum window Seal replacement
Five Killer Quora Answers To Link Togel Resmi link Togel
The Most Inspirational Sources Of Leather Sofas Near Me leather corner couch (chordgrain06.bravejournal.Net)
5 Killer Quora Answers On Composite Door Frame Replacement composite door frame replacement
15 Top Pinterest Boards From All Time About Upvc Replacement Window Handles upvc windows repair near me [Wiley]
5 Laws That Can Help The Adhd Private Assessment
Industry Adhd Assessment Scotland
Why Stress And Anxiety Symptoms Isn’t A Topic That People Are Interested In Stress And Anxiety Symptoms anxiety hangover symptoms (Merle)
10 Methods To Build Your Cost To Replace Windows Uk Empire Replacement glass for window near me
5 Laws That Can Help The Mesothelioma Asbestos Industry Mesothelioma lawyer
How To Determine If You’re In The Mood To Adult Adhd Symptoms Women adhd
disease Symptoms [https://Iampsychiatry07584.designertoblog.com/60440505/the-myths-and-facts-behind-adhd-symptoms-In-women-test]
5 Clarifications Regarding Sash Window Refurbishment bespoke sash windows
Five Things You’ve Never Learned About How To Get An ADHD Diagnosis adhd Diagnosis and stigma
Who’s The World’s Top Expert On Couch L Shaped Sofa? l shape sofa bed (advicebookmarks.com)
What’s The Job Market For Window Doctor Near Me Professionals?
window doctor
The Ultimate Glossary Of Terms About Asbestos Cancer Lawyer Mesothelioma Settlement Asbestos lawyer
10 Myths Your Boss Has Regarding Situs Togel Online Situs Toto Togel
The symptoms can appear 10 to 50 years after initial exposure and often mistakenly identified as other
illnesses such as influenza or pneumonia.
Here is my webpage – Asbestos Legal
20 Great Tweets Of All Time About ADHD Private Assessment Adhd assessment Private uk
This Is The Myths And Facts Behind Electric Fireplace Wall Mounted
modern fireplace (Lamar)
What’s The Current Job Market For ADHD Titration Waiting List Professionals Like?
Adhd titration waiting List
10 Mobile Apps That Are The Best For L Shaped Triple Bunk Bed Double And Single Bunk Beds
Are How To Get An ADHD Diagnosis The Most Effective Thing That Ever Was?
getting an adhd diagnosis Uk
The 10 Most Terrifying Things About Underdesk Treadmill underdesk treadmill,
netvoyne.ru,
A Provocative Rant About Replacement Window Handle upvc sliding door handle (Janessa)
10 Meetups About ADD And Treatment You Should Attend adhd in Adults untreated
15 Unexpected Facts About Nissan Car Key Replacement
That You Didn’t Know About nissan figaro key (forexmob.ru)
Why No One Cares About Pragmatic Free 프라그마틱 환수율
Nine Things That Your Parent Teach You About Bi-Fold Door Repair
bi-Fold door repair; clashofcryptos.trade,
17 Reasons Not To Ignore Walking Pads For Under Desk running
Guide To Situs 4d: The Intermediate Guide To Situs 4d situs
4d (Georgetta)
Find Out More About Sectional Sleeper When You Work From At Home sleeper Sectional Couch
How To Beat Your Boss With Bentley Key Fob Bentley car keys
What’s The Job Market For Double Glazed Window Repairs Near Me Professionals Like?
Window repairs near me
15 Terms That Everyone Working In The Double Glazing Windows
Repairs Industry Should Know contractors
This Is A Audi A4 Key Replacement Success Story You’ll Never Believe
replace audi key fob [penelopetessuti.ru]
9 Things Your Parents Teach You About Treadmills With Incline
For Sale treadmills With incline for sale
Saab 93 Key Fob Replacement Explained In Less Than 140 Characters Saab Key Fob Replacement
The Three Greatest Moments In Case Battle History case battle cs go (maps.Google.com.Qa)
20 Trailblazers Are Leading The Way In Audi Keys audi A3 Spare key
Five Killer Quora Answers On Link Togel Resmi link togel resmi (https://www.metooo.com/u/66cdd8a1c2c560701e8fee6f)
Why Do So Many People Want To Know About Pragmatic Genuine?
무료슬롯 프라그마틱
It Is The History Of Skoda Yeti Key skoda kodiaq key (Alannah)
Why Single Bunk Will Be Your Next Big Obsession bunk beds two single
(Stacie)
The 10 Most Scariest Things About Volvo Keyfob Volvo s60 key
You’ll Never Guess This Three Wheel Pushchair’s Benefits Three Wheel
Pushchair (https://Www.Bitsdujour.Com/)
10 Meetups On Electric Fire Suite UK You Should Attend electric fireplace Freestanding
Learn About Citreon Key While Working From Your Home unlock car door
The 10 Scariest Things About Bifold Door Seal Repair bifold door seal
Repair (lovewiki.faith)
Responsible For An Electric Treadmill Treadmill Budget? 10 Terrible Ways To Spend Your Money slim Electric
Treadmill (Peatix.com)
The 10 Most Scariest Things About Mesothelioma Lawyer mesothelioma (Emery)
Why We Love Electric Fire Wall Mounted (And You Should Also!) fireplace wall mounted, Joey,
5 Qualities That People Are Looking For In Every Online
Mystery Box the best mystery boxes
Distributing Your Internet Marketing Message With Podcasts
주소모음
Choosing Suitable Domain 주소모음
Setting Up A Web Page The Right Way 주소모음
Building Website That Your Visitors Will Love 주소주라
Linking Strategies – Construction Traffic On Your Own Web Site
주소모음 (http://hmhcwin.com)
Basic Seo Tips That Can Help You Promote Internet Site 링크모음
(doc1000.hungerfordprimaryschool.co.uk)
Seo Techniques And Service Providers 링크모음
15 Strange Hobbies That Will Make You More Effective At Audi A4 Key Replacement replace audi key fob
3 Wheel Pushchairs Isn’t As Difficult As You Think 3 Wheel twin Stroller
Link Building Tips For Obtaining Relevant Backlinks 주소모음
Home Cartomancy Business – A Love Story 주소주라
The 10 Scariest Things About Car Key Cutting Price Car key cutting price
Internet Marketing – The Two Secret Words 주소모음
11 “Faux Pas” That Are Actually OK To Make With Your Cot
Beds Cot Beds For Sale
Online Best Seller Publishing: How To Start With It? 주소모음
Social Bookmarking – Going Where The Traffic Is 링크모음
15 Top Twitter Accounts To Learn About Audi Replacement Key programming
5 For You To Make Google Crawl Have A Lot Site 주소주라
Measure Roi Of Constructing 링크모음
A Look At The Future What’s The Buy Butt Plug Industry Look Like In 10
Years? sex and Butt plug (hatcher-lorentsen.technetbloggers.de)
Unexpected Business Strategies For Business That Aided Private Testing
For ADHD Succeed private adhd assessment coventry
Submit New Website Url And Sitemap To Google, Yahoo, And Bing 링크모음
15 Best SEO Company Bloggers You Should Follow Best Seo Company
An Easy-To-Follow Guide To Pragmatic Official Website 프라그마틱 데모 (http://90pk.com/home.php?mod=space&Uid=372954)
Software SEO Tools To Ease Your Daily Life Software SEO Trick That Every Person Must Be Able To Software Seo
10 Key Factors About Sectional With Chaise And Sleeper
You Didn’t Learn In The Classroom sectional Sofas with hide a bed
The 10 Most Scariest Things About Accident And Injury Lawyers accident And injury lawyers
20 Incline Treadmill Websites Taking The Internet By Storm portable treadmill
incline – Nichol –
A Journey Back In Time What People Said About Repair Double Glazing 20 Years Ago
double glazed window Repairs
10 Of The Top Mobile Apps To Use For Rolls Royce Car Key repaired
Cat Flap Glass Door Installation Near Me cat flap installation
Simple To Help Make Money Online 링크모음
How These Semi-Automated Submitting Of Social Bookmarks Submitters Help You
To You 주소주라
One Of The Biggest Mistakes That People Make With Hire Car
Accident Lawyer lawyer car accident Near me (https://www.hulkshare.com)
What Is The Case Battles Term And How To Use It Case Battle Win
11 Ways To Completely Redesign Your Private ADHD Assessment UK adult adhd private assessment (Tina)
Why Misted Double Glazing Repairs Can Be More Risky Than You Think
repairing double glazing
The Leading Reasons Why People Perform Well With
The Replacement Car Key Vauxhall Industry car key cutting and programming price
Question: How Much Do You Know About SEO Company Manchester?
best Seo company in Uk
Internet Traffic – Better Alternative To Link Wheels 주소모음
Five Essential Qualities Customers Are Searching For In Every Cot Bed cheap baby cots for sale (http://www.daoban.org)
10 Things Everyone Gets Wrong About The Word “Pragmatic.” 프라그마틱 사이트
You’ll Be Unable To Guess Genuine Ferrari Replacement Key Uk’s
Secrets genuine ferrari replacement key uk
Responsible For An Nissan Key Fobs Budget? 10 Wonderful Ways To Spend Your Money
Nissan key Cut
The Underrated Companies To Follow In The Bio Fuel Fire Industry bio fuel Fireplaces
What The Heck What Is Online Sport Toto? 토토사이트 추천 (http://Ckxken.Synology.Me/Discuz/Home.Php?Mod=Space&Uid=295511)
See What New Ferrari Key Fob Tricks The Celebs Are Utilizing new ferrari Key fob
How You Can Use A Weekly Wall Mounted Electric Fireplace Project Can Change Your Life White fireplace
You’ll Never Guess This 3 Wheel Compact Stroller’s Secrets 3 wheel compact stroller
17 Reasons To Not Not Ignore Bmw Replace Key replace
bmw key (Poundhubcap8.bravejournal.net)
How Fold Treadmill Changed My Life For The Better foldable treadmills (Mac)
A Relevant Rant About Curved Couch Sofa Couch Deals (Carpberet7.Werite.Net)
See What Sectional L Shaped Sectional Tricks The Celebs Are Making
Use Of sectional l Shaped Sectional
The Worst Advice We’ve Received On Adhd Assessment For Adults
why are adhd assessments so expensive
The 10 Scariest Things About Pragmatic Sugar Rush 프라그마틱 체험
Why Nobody Cares About French Door Refrigerator With Ice Dispenser
french door fridge freezer australia
Scuttle For Social Bookmarking: Is It Worth
Period? 주소주라, yanagawa-sports-seikotuin.com,
15 Inspiring Facts About Item Upgrade That You’ve Never Heard Of upgrade items (Kasey)
How Drugs Simple Social Websites Stronger For Your
Targeted Site 링크모음
4 Crucial Hubpages Seo Tips For Your Personal Published
Web Page 주소모음
The Top Blue Sectional Sofa Gurus Do Three Things
green sectional Sofa (https://highwave.kr/bbs/board.php?bo_table=faq&wr_id=2873851)
Paid Online Survey – Paypal Once Again Gets You Paid Cash Fast For
Doing Surveys 링크모음 (https://www.google.com.au/)
Five Quick Tips To Get Your Blog On Top 10 Of Google 링크모음
Guide To Upvc Patio Door Hinges: The Intermediate Guide In Upvc
Patio Door Hinges Upvc Patio Door Hinges
Ten Things Your Competitors Inform You About
UK Private ADHD Diagnosis private adhd Assessment Durham
7 Simple Tips For Moving Your Mystery Box online mystery box websites
9 Things Your Parents Teach You About Green Velvet Chesterfield Sofa green velvet chesterfield sofa (Jenni)
What’s The Current Job Market For How Much Fabric For 2 Seater
Sofa Professionals Like? how much fabric for 2 seater sofa – Augustus,
20 Myths About Audi A3 Key Battery: Dispelled replacement car key audi
14 Misconceptions Commonly Held About ADHD Private Assessment private adhd assessment harrogate [https://bookmarkshome.com/story3437954/10-facts-about-adhd-private-assessment-that-can-instantly-put-you-in-the-best-mood]
What Truck Accident Lawyers Near Me Experts Would Like
You To Know trucking accident injury lawyer – algowiki.win –
Guide To Locksmith Near Me Prices: The Intermediate Guide For
Locksmith Near Me Prices locksmith near me Prices
Five People You Must Know In The Item Upgrades Industry item upgrader Mod
20 Inspirational Quotes About Suite Electric Fireplace Fireplace Electric With Mantel,
https://Davidson-Yu.Thoughtlanes.Net/11-Creative-Ways-To-Write-About-Fireplaces-Electric-Suites,
10 No-Fuss Methods For Figuring Out Your Upvc Door And Windows Double Glazing Upvc Windows
Since mesothelioma has a long latency period, it could take decades for symptoms
to begin to manifest. A mesothelioma lawyer will examine medical records, look into the history of work, and collect evidence to build a strong case.
Also visit my site – Asbestos Litigation (Oakley-Hawkins.Thoughtlanes.Net)
14 Cartoons About Audi Car Key Replacement Which Will Brighten Your
Day Audi A3 Key
Mesothelioma is a deadly form of cancer caused exclusively by exposure to asbestos lawsuit;
Xavier,
. A New York mesothelioma lawyer can assist victims and their families in holding companies accountable for concealing this dangerous substance.
8 Tips To Increase Your Pragmatic Demo Game 프라그마틱 슬롯 사이트 (Emory)
This Week’s Top Stories About Mesothelioma And Asbestos Lawyer Mesothelioma And
Asbestos Lawyer Mesothelioma attorney
What’s Holding Back The Wall Electric Fireplace Industry?
modern electric fires wall mounted (Bailey)
Ten Sports Totos That Really Improve Your Life 토토사이트 순위 (Lonny)
15 Things You Didn’t Know About Upgrade Item item upgrader mod
(Brittney)
You’ll Never Guess This Window Replacement Near Me’s
Tricks window replacement near Me
Adults Bunk Bed Explained In Fewer Than 140 Characters Bunk Beds usa
Guide To Leather Chesterfields For Sale: The Intermediate Guide To Leather Chesterfields For Sale leather chesterfields
for sale [Gail]
10 Things That Your Family Taught You About Good Cribs good Cribs
15 Gifts For The Fireplace Surround Lover In Your Life marble fireplace
10 Things That Your Family Teach You About ADHD Assessment Adults Uk adhd assessment adults uk (Geneva)
Three Greatest Moments In Car Key Repair History Car Remote Key Repair Shop Near Me
Here’s A Little Known Fact Regarding 5kw Wood Burning Stove Outdoor Wood Burning Cookers, https://Clicavisos.Com.Ar/Author/Collarman9,
What Causes Mesothelioma Other Than Asbestos Explained In Fewer Than 140 Characters Mesothelioma Attorney
The Volkswagen Car Key Replacement Mistake That Every Newbie Makes Volkswagen Lost Keys
From Around The Web From The Web: 20 Awesome Infographics About Used Wood Burning Stoves Portable wood fireplace
10 Places Where You Can Find Americanfridge Freezer American Fridge Freezers For Sale Uk – Timeoftheworld.Date,
The 10 Most Terrifying Things About Four Seater Chesterfield Sofa four seater chesterfield
sofa (Alannah)
Electric Fireplace Mantels’s History History Of Electric Fireplace Mantels Biofireplace
The Best Way To Explain Wood Stove Burning To Your Mom large Wood burner (Netvoyne.ru)
Key Programmers It’s Not As Hard As You Think car key cut and programed (Jonathan)
How To Tell If You’re Prepared For Replacement Audi
Car Key audi smart key replacement
See What Pellet Stoves For Sale Near Me Tricks The Celebs Are Using pellet stoves for sale near me
What Is Pragmatic Return Rate And Why Is Everyone Speakin’ About It?
프라그마틱 무료 슬롯버프
15 Things You’ve Never Known About Cheap Leather Couches leather sofas for sale (Kraig)
See What Ferrari Replacement Key Cost Uk Tricks The Celebs Are Making Use Of
ferrari replacement key Cost Uk
15 Gifts For The Fireplace Surround Lover In Your Life marble
fireplace (Conrad)
Twenty Myths About Pushchair 3 Wheels: Busted 3 wheel baby stroller;
https://humanlove.stream/wiki/jensbyjohannsen8168,
A Provocative Rant About Double Glazed Windows Bromley Bromley door company (http://www.airsoftmarkt.Nl)
You’ll Never Guess This Link Building Management Software’s Tricks link building management software
5 Laws That Will Help Industry Leaders In Replacement
Key For Audi Industry Car key cover audi
“The Glass Doctor Awards: The Most, Worst, And The Most Bizarre Things We’ve Seen window seal replacement
5 Reasons To Be An Online ADHD Medication And 5 Reasons Why You
Shouldn’t online adhd medication, Samuel,
20 Resources To Make You More Effective At Item Upgrade
item level upgrade [Theflatearth.Win]
Everything You Need To Know About Double
Glazing Sealed Unit Replacement replacement sealed window units [Historydb.Date]
What A Weekly Car Key Cut And Programed Project Can Change Your Life
Car key replacement
15 Interesting Facts About Train Accident Cases That You’d Never Been Educated About lawyer truck accident
Guide To Childs Pram: The Intermediate Guide The Steps To Childs Pram Childs Pram
– Rubigordon.Com,
You’ll Never Be Able To Figure Out This Car Key Fob Repair Near Me’s
Tricks key fob repair near Me – perfectworld.wiki –
10-Pinterest Accounts You Should Follow About Best Budget Espresso Grinder coffee grinder materials
20 Quotes That Will Help You Understand Oxblood Chesterfield Leather Two Seater Chesterfield
Why We Love Fold Flat Treadmill (And You Should, Too!) Treadmills that fold flat
Watch Out: How Case Opening Battles Is Taking Over And What Can We Do About It case battle win (Janeen)
What Is It That Makes Coffee Beans Machine So
Popular? coffee bean to cup machines (Metooo.co.uk)
5 Reasons Best French Style Fridge Freezer Is Actually A Great Thing french door fridge
The Best Subcold Mini Fridge Strategies To Transform Your Life Cheap Refridgerators
Why You Should Focus On Making Improvements In Locksmith Near Me House near Me Locksmith
Backlink Tool: Fun Setting Up Ideas 링크모음
A Look At The Ugly The Truth About Private Psychiatrist Liverpool private psychological Evaluation
Are Link Wheels Effective As A Seo Scheme?
주소주라, http://www.tzro.cc,
20 Top Tweets Of All Time About Hyundai Spare Key replacement key fob near me
15 Best Item Upgrade Bloggers You Must Follow Best item Upgrade
Sectional Sleeper Sofa Tools To Streamline Your Daily Lifethe One Sectional Sleeper Sofa Trick That Should Be Used By
Everyone Know sectional Sleeper sofa, livebookmarking.com,
The Largest Issue That Comes With Pragmatic Official Website, And How You Can Repair It 프라그마틱 슬롯 사이트 (https://www.google.com.co/url?q=https://squareblogs.net/weaponknight1/how-much-can-pragmatic-slot-experience-experts-earn)
Tips On How To Use Delicious For Social Bookmarking 링크모음
10 Amazing Graphics About Case Battles Case battle Cs2
Are You Planning To Outsource Website Link Building Is Most Effective?
주소주라
10 Signs To Watch For To Get A New Item Upgrader Item Upgrading
How To Get More Benefits Out Of Your Maxi Cosi Stroller And Car Seat maxi cosi mico car seat [Julio]
7 Tips About Folding Electric Treadmill That Nobody Will Share
With You Portable Electric Treadmill [Ovesen-Urquhart.Blogbright.Net]
20 Things Only The Most Devoted Best Bunk Bed Fans Understand best bunk beds with storage
Bookmark Marketing Made Simple 주소주라
Lost Mitsubishi Car Keys Techniques To Simplify Your Daily Lifethe One Lost Mitsubishi Car Keys Trick That Every Person Must Be
Able To lost Mitsubishi car Keys
5 Killer Quora Answers To Mobility Scooter For Outdoor Use Mobility Scooter For Outdoor Use
20 Up-And-Comers To Watch In The Case Battle Industry cs case
battle (http://enbbs.instrustar.com/)
The Next Big New Mesothelioma Asbestos Claim Industry mesothelioma lawyers – https://valetinowiki.racing/wiki/Five_Lawsuits_Mesothelioma_Lessons_From_The_Pros –
You Can Explain Case Opening Battles To Your Mom case opening battle csgo
What Is Coffee Machines With Pods And How To Utilize It?
pod coffee maker
Why Nobody Cares About Mesothelioma Compensation mesothelioma lawyers (Anh)
Guide To Treadmill Fold Up: The Intermediate Guide The Steps
To Treadmill Fold Up Treadmill Fold Up
The Companies That Are The Least Well-Known To Watch In The
Upvc Doors Repair Industry upvc door frame Repair
The Best Advice You’ll Ever Receive On Car Key Fob Repair car
Key fob repairs near me; Nutris.net,
20 Irrefutable Myths About Audi Spare Key: Busted cost of audi key replacement
The Ultimate Glossary On Terms About Mystery Box mystery box best
15 Best Pinterest Boards Of All Time About Toto Result 스포츠토토 사이트
10 Misconceptions Your Boss Shares About Online Mystery Boxes online mystery box Opening
7 Useful Tips For Making The Most Out Of Your Private ADHD Assesment private adhd asd assessment
Near Me (top10bookmark.com)
Ninja Back Link Building Part Six – Creating Of Social
Bookmarks 링크모음
You’ll Never Guess This Renault Zoe Key’s Tricks
renault Zoe Key
Find Out What Auto Locksmiths Near Me Tricks Celebs Are Making Use Of 24 Hour Locksmith Auto
Do You Think You’re Suited For Doing Case Battle?
Answer This Question cs case Battle
You’ll Never Guess This Wall Mounted Bio Ethanol Fireplace’s Tricks Ethanol fireplace
Crossing The Streams – Top 5 Ways Encourage Your World Wide Web Offline 주소주라
This Is The Myths And Facts Behind Couch With Chaise Lounge Chaise lounge sofa
The Most Hilarious Complaints We’ve Seen About Upvc
Window Repairs window repairs near Me
Using An Effective Backlink Campaign For Your Online
Business 검색엔진최적화 seo
25 Unexpected Facts About Accident Attorney Accident attorney near me
A Simple Guide To Analyzing Many Of Those Web Traffic Ideas – Part 4 링크모음
Top 3 Tips To Bookmark Marketing 주소모음
How Acquire Ownership Of One’s Affiliate Link Or Replicated Website 주소모음
Swan Retro Fridge Freezer Cream Tools To Ease Your Daily Life Swan Retro Fridge Freezer
Cream Trick That Everybody Should Know Retro Fridge Freezer
The Most Prevalent Issues In Pragmatic Genuine 프라그마틱 무료게임
Your Worst Nightmare About Online Mystery
Box Relived Best online mystery box site
The Little Known Benefits Of Fabric 2 Seater Sofa Small fabric 2 seater sofa
Be On The Lookout For: How Pragmatic Slots Experience Is
Taking Over And What Can We Do About It 프라그마틱 데모
5 Clarifications On Car Accident car crash lawyers
5 Killer Quora Answers On Arabica Coffee Beans beans (Hildegard)
The Best Way To Explain ADHD Diagnosis Near
Me To Your Boss adhd private diagnosis Cost uk
The Most Underrated Companies To Follow In The Folding Scooters Industry 26 Stone Folding Mobility Scooter
The Top Companies Not To Be Keep An Eye On In The Commercial Espresso Machine Industry Home espresso machine
Do Not Believe In These “Trends” Concerning Pragmatic
프라그마틱 정품 사이트, Bookmarkassist.Com,
The Biggest Problem With Good Accident Lawyers, And What You Can Do To Fix It Good Accident Lawyers Near Me, Elearnportal.Science,
10 Misconceptions Your Boss Holds About Auto Accident Compensation Auto Accident
Compensation Car injury lawyer near me (Forums.indexrise.com)
10 Mistaken Answers To Common Semi Truck Accident Questions:
Do You Know The Right Ones? Semi truck Accident Lawyers
Guidelines For Link Building 주소주라
Nine Things That Your Parent Teach You About Two Seater Fabric Sofa Two seater Fabric Sofa
8 Tips To Boost Your Trucking Accident Lawyers Game semi truck accident attorney near me (clashofcryptos.trade)
15 Shocking Facts About Industrial Door Repairs Cambridge door seal repair near me
Ten Things Your Competitors Teach You About Item Upgrades Upgrade item
The Ultimate Glossary Of Terms About Mesothelioma Attorney mesothelioma litigation, Loretta,
A Look In Ferrari Key Replacement Near Me’s Secrets Of Ferrari Key Replacement Near Me ferrari remote key
Why No One Cares About Renault Clio Key Card Replacement new key for renault clio (maps.google.com.sl)
The History Of Car Key Cutting Cost key Duplication
14 Common Misconceptions Concerning Case Battles case battle cs go – Edwin,
9 . What Your Parents Taught You About Mesothelioma Claim mesothelioma
Are Case Opening Battles Just As Important As Everyone Says?
Csgo battle case
“Ask Me Anything:10 Answers To Your Questions About Adhd Assessment London how Do
i get an Adhd assessment
13 Things About 3 Wheel Buggy You May Not Know 3 Wheel Stroller With bassinet
10 Tell-Tale Warning Signs You Should Know To Get A New Grey Chesterfield Corner Sofa
grey fabric Chesterfield sofa
Top Ten Tips For Marketing A Questionable Income Scheme Offline 링크모음
Choosing Effective Domain Name 링크모음
You’ll Never Guess This Seat Ibiza Key Fob Replacement’s Tricks
seat ibiza Key fob replacement
You’ll Be Unable To Guess Best Cots For Newborns’s Tricks best cots for newborns; Corine,
15 Things You’ve Never Known About Sash Double Glazing sash window renovation
10 Misconceptions Your Boss Has About Reprogramming Car Key Reprogram key fob
The Most Pervasive Issues With Treehouse Bed Play Area Bunk Beds
What Will Fiat 500 Replacement Key Cost Be Like In 100 Years?
fiat tipo Key
5 Best G Spot Toy Lessons From Professionals best G spot
toys, Valetinowiki.racing,
The Best Single Buggy Methods To Transform Your Life cheap single Buggy
5 Killer Quora Answers To Strollers 3 Wheels Strollers 3 Wheels
The Hidden Secrets Of Double Glazing Doors Repairs double glaze window repair
Internet Marketing – A Technique 주소모음
How Even Worse Photo Bookmarks 주소모음
16 Must-Follow Facebook Pages For Electric Fire Suites Oak Marketers slim electric Fireplace with mantel
Car Key Replacements Near Me Tools To Streamline Your Daily Lifethe One Car Key Replacements Near Me Technique Every Person Needs To Be Able To car key replacements near Me
9 Lessons Your Parents Taught You About Treadmill Electric
Motor treadmill electric motor (Margarette)
A New Way Of Link Crafting? 링크모음
Internet Explorer Tips That Improve Your Internet Surfing Experience 주소주라
The Link Swapping Trap 링크모음
A Quick, Free, And Guaranteed Way Of Getting A New Web
Site Indexed By Google 주소모음
Money Making Ideas – Make Money Online Without Your Own Product!
링크모음
Ten Programming A Car Keys That Really Make Your Life Better keys
17 Reasons Why You Shouldn’t Be Ignoring Volvo Replacement Key volvo Replacement Keys
Why Is Psychiatric Assessment Private So Famous? Urgent Psychiatric Assessment – Explorebookmarks.Com –
Why 2 Seater Chaise Should Be Your Next Big Obsession 3 seater chaise lounge (Foster)
It’s The Ugly Truth About Pragmatic Free Slots 프라그마틱 슬롯 추천
Guide To Replacement Key For Audi: The Intermediate Guide For Replacement Key
For Audi Replacement key for audi
10 Things You’ve Learned About Preschool That Can Help You In Link Togel togel sydney
How To Join Up To A Site And Connect It To Web Site 링크모음
Reciprocal Link Deception 주소모음
Wedding Favors – Beach Gifts To Thrill Your Guests 주소주라
10 Mistakes To Avoid When Setting Up A Link Directory 주소모음
Market Business Online: 3 Hot Tips! 주소모음
Upvc Patio Door Hinges: A Simple Definition double glazing window hinge repairs
What’s The Job Market For Foldable Wheelchair Uk Professionals Like?
Foldable wheelchair uk
The Top 5 Reasons People Thrive In The Audi Replacement
Key Industry audi Q7 key
The Reason Behind Volvo Replacement Key Fob Is Everyone’s Obsession In 2023 volvo xc90 remote start key fob, https://bysee3.com/home.php?Mod=space&uid=4379362,
The Good And Bad About Pragmatic 무료슬롯 프라그마틱
How To Beat Your Boss On Pram 2 In 1 2 in 1 baby
Pram (images.google.ms)
10 Things That Your Family Teach You About Undiagnosed ADHD In Adults diagnosed Adhd
Looking For About A Reverse Directory Lookup? 주소주라
Item Upgrading Explained In Fewer Than 140 Characters Upgrade Item
It’s The Ugly The Truth About Online Mystery Box best one Time mystery box
10 Real Reasons People Dislike Mesothelioma Lawsuits Mesothelioma Lawsuits Mesothelioma claim
Ten Male Masterbating Toys Myths That Aren’t Always True male masturbation devices
10 Life Lessons We Can Take From Online Mystery Box the best mystery boxes
Make Money Using Adsense – Learn How Using Link Units Can Increase Your Adsense Revenue
주소주라
See What Fresh Ground Arabica Coffee Beans Tricks The Celebs Are Using fresh ground Arabica coffee beans
The Abcs Of Generating 1 Way Links To Raise Your Search Engine Placement 주소주라
Guide To Accident Injury Lawyers: The Intermediate Guide To Accident
Injury Lawyers Accident Injury lawyers (valetinowiki.Racing)
20 Trailblazers Are Leading The Way In Private ADHD Clinic
Private ADHD Assessment Brighton cost (https://socialmediaentry.com/)
Five Killer Quora Answers To Cars Keys Replacement Cars
Keys Replacement (https://Lovebookmark.Date/)
15 Documentaries That Are Best About Pragmatic Experience 무료슬롯 프라그마틱 (https://naturalbookmarks.com/story18098339/a-step-by-step-guide-for-choosing-the-right-pragmatic)
5 Ultra Effective Marketing Strategies Improve Your Company 링크모음
Internet Marketing Strategies For Success Online 링크모음
(timemapper.okfnlabs.org)
Cheapest Anal Toy: What’s The Only Thing Nobody Is Talking
About anal sex toys
Say “Yes” To These 5 Toto Tips 안전놀이터
Ten Accident Injury Attorney Myths You Shouldn’t Share On Twitter accident attorneys
10 Undeniable Reasons People Hate Single
Standing Stroller single seat Stroller
Bandar Online Togel Tools To Ease Your Daily Lifethe One Bandar Online Togel Trick Every Individual Should Be Able To bandar online togel, Christin,
Ten Things Everybody Is Uncertain About Programing Car Keys
Key programming
20 Trailblazers Leading The Way In Sport Toto Website 먹튀검증 사이트 (Anja)
Why Adding Toto Online Terbaik To Your Life Will Make All The
Impact Bandar Toto
Fabric Cream Corner Sofa Tools To Streamline Your Everyday Lifethe Only Fabric Cream Corner Sofa Trick
Every Individual Should Be Able To fabric cream Corner Sofa
Ezine Marketing – Easy Methods To Triple Your Ezine Opt-Ins With A Squeeze Page 링크모음
9 . What Your Parents Taught You About Accident Lawyer Near
Me accident lawyer near me (Humanlove.stream)
Guide To How Much Is A Private ADHD Assessment: The Intermediate Guide On How Much Is
A Private ADHD Assessment how much is a private Adhd assessment
15 Things To Give Those Who Are The Double Glazing Repairs Luton Lover In Your Life windows And Doors near me
What Is ADD Natural Treatment? History Of ADD Natural Treatment Drugs To Treat Adhd
The 10 Most Terrifying Things About Window Hinge Repair Near Me Window Hinge repair near Me
5 Laws That Anyone Working In Item Upgrading Should Know upgrade item [Adrienne]
Who’s The Top Expert In The World On Audi A3 Replacement
Key? reprogram
5 Killer Quora Answers On Lamborghini Key Replacement Cost lamborghini Key Replacement
5 In Order To Link Building Success 주소모음
The 3 Biggest Disasters In Private Psychiatrist South Wales The Private Psychiatrist South Wales’s 3 Biggest Disasters In History Private Psychiatrists Uk
20 Trailblazers Leading The Way In Emergency Locksmith Service Near Me local emergency locksmith
See What Bmw 1 Series Key Tricks The Celebs Are Using Bmw 1 Series Key
The Ultimate Cheat Sheet On Mystery Boxes Mystery Box Opening Online (https://Images.Google.Cg)
10 Wrong Answers To Common Commercial Espresso Machine Questions:
Do You Know The Right Answers? espresso capsule machine
Social Bookmark Tips For Rookies 링크모음 [http://ultfoms.ru/user/FloydDugger186]
The Reasons To Focus On Enhancing Replace Window Handle Cost Of Windows Replacement
What Do You Know About Cheap Leather Couches? leather sofas
for sale near me (Rosalina)
15 Shocking Facts About Car Key Cutting Machine That You Never Known Auto key Cutting
Car Key Programing Tips From The Top In The Industry
Keys Programmed
How To Find The Perfect Private ADHD On The Internet Adhd Assessment Scotland Private
10 Facts About Bmw Replacement Keys That Make You Feel Instantly A Good Mood Keys for Bmw
Why Pragmatic Slots Site Is Fast Increasing To Be The Hot Trend For 2024?
프라그마틱 슬롯 무료체험
Small Url – Hiding Your Affiliate Links Without Spending
A Dime 주소모음
The 10 Most Terrifying Things About Wood Burner Fireplace Ideas wood burner
fireplace; Emelia,
What NOT To Do In The Mesothelioma Lawyer Industry Mesothelioma Law firms
The Best Item Upgrade Tricks To Change Your Life Best Item Upgrade
How To Make Money On The Online World – Easy 주소주라
Do You Think Good Accident Lawyers Ever Be The King Of
The World? good accident lawyers near me
Nine Things That Your Parent Taught You About Pellet Stove Stores Near Me pellet Stove stores near me (https://lake-rosenkilde-2.federatedjournals.com/)
5 Killer Quora Answers To Composite Door Hinge Replacement
Composite Door Hinge Replacement
Effective Ways In The Particular Top Url Of Your Website
For Blog Site 주소주라
7 Tips About Asbestos Attorneys Oklahoma That Nobody Will
Tell You mesothelioma Lawyers
You’ll Never Be Able To Figure Out This Large
2 Seater Fabric Sofa’s Benefits large 2 Seater fabric Sofa
See What Double Glazing Repairers Tricks The Celebs
Are Utilizing double glazing repairers (Trent)
The No. 1 Question Anyone Working In Situs Togel Terpercaya Must Know How To Answer Togel Resmi Indonesia
24 Hours For Improving Lawyers For Accident lawyer near me
accident (Celinda)
Technology Is Making Key Programmer Better Or Worse?
How Much To Reprogram A Car Key Uk
The Reasons You’re Not Successing At Cut Car Key Near Me van key cutting – Alisa –
What’s The Current Job Market For Accident Claims Lawyers Professionals Like?
Accident Claims Lawyers
Reallife Sex Dolls Tips To Relax Your Daily Lifethe One Reallife Sex Dolls Trick That Every Person Must Learn reallife sex dolls (Kala)
Why Repair Upvc Windows Could Be Your Next Big Obsession? screwfix upvc windows
The 10 Most Terrifying Things About Sports Toto Result Latest 먹튀검증 사이트 (Launa)
9 . What Your Parents Teach You About Leather Sectionals For Sale
leather sectionals For sale
What You Can Do To Get More From Your ADHD Diagnosis
Private UK Adhd assessment for adults private
10 Tips For Getting The Most Value From Truck Accidents Lawyer truck accident attorney
(planforexams.Com)
Social Media Does Not Mean Running For The Hills 링크모음 (http://namheui.kim/)
The 10 Most Terrifying Things About Senseo Philips Coffee Maker
senseo Philips coffee maker
Does Your Facebook Fanpage Suck? Teach These Tips
To Really Hot! 링크모음
What’s The Good And Bad About Mesothelioma Legal Question mesothelioma claims (Noah)
20 Fun Details About Triple Sleeper Bunk Bed three bed bunk bed (Rosella)
Guide To Situs Togel Dan Slot Terpercaya: The Intermediate Guide In Situs Togel Dan Slot
Terpercaya situs togel dan slot terpercaya
The Best Integrated Fridge Freezer Is Gurus. Three Things
50/50 fridge freezer built in (https://cameradb.review)
On-Page Seo Can Increase Your Page Ranking 주소모음 – kimaarkitektur.no,
Guide To Private ADHD Assessment Online: The Intermediate Guide
The Steps To Private ADHD Assessment Online Private adhd assessment online
The 10 Most Terrifying Things About Mens Masturbators Mens Masturbators
The 10 Scariest Things About ADHD Private Assesment adhd assessment leeds Private
Bookmarking – Droid Tips That Work 주소모음 (http://www.philharmony.com)
The 10 Most Terrifying Things About Adult Adhd Assessments
adhd assessments (https://bookmarkcolumn.com)
4 Most Frequently Found Ways To Promote Your
Affiliate Url 주소모음
25 Surprising Facts About Coffee Machines Best which coffee machines
are best, Bobby,
See What Keys Programmed Tricks The Celebs
Are Making Use Of keys Programmed
The 12 Most Popular Asbestos Mesothelioma Lawyers Accounts To Follow On Twitter mesothelioma lawsuits – http://www.Metooo.Es,
How To Add Url To Metacrawler 주소모음
Why Porsche Macan Key Isn’t A Topic That People Are Interested In Porsche Macan Key porsche replacement key Fob
What Is Fire Pits & Chimineas And Why Is Everyone Dissing It?
Chimineas And Fire Pits
5 Oneway Link Building Tips For Your Serious Marketer 주소주라
Ten Lightweight 3 Wheel Strollers That Really Help You Live Better 3 wheel strollers (taikwu.com.tw)
10 Apps To Help You Control Your Mesothelioma mesothelioma compensation
Make Money Online Essentially 5 Simple Methods 주소주라
Psd To Html – How To Make It Worse Your Website Search Engine Friendly 링크모음; https://cse.google.com.eg/,
How To Conduct Link Campaigns – And Make A Success! 링크모음
Buy Traffic — 5 Ways To Instantly Increase Site Visitors 주소주라
Internet Marketing Tips – U Is Right For Url
링크모음
Importance Of Experiencing A Website For Your Online 주소주라 (google.co.za)
I know this web site offers quality dependent articles or reviews and
other stuff, is there any other web page which presents these stuff in quality?
Web Hosting Domain Names Packages 주소주라; bio.rogstecnologia.com.br,
Four Outdated Link Building Strategies 링크모음
The Reality Of Yahoo Submissions 주소주라
Search Engine Marketing And Link Building – Will Way 링크모음
Seo Tlc For Latest Web Site 링크모음 (http://www.lsbin.com)
Getting Cyberduck And An Ftp Connection Set Till Upload Website 주소모음 (http://m.xqygm.scenery.co.kr/)
6 Quick Steps Different More Money With Link Popularity 링크모음
How A Weekly Best SEO Software Project Can Change Your Life Seo Software For Ranking Tracking (https://Www.Longisland.Com/)
What’s The Job Market For Best Oven Uk Professionals?
Best Oven Uk
10 Unexpected Masturbator Tips best male masterbaiters, Rafael,
You’ll Be Unable To Guess Anal Sex Toy Price Uk’s Tricks anus sex Toy
What’s The Current Job Market For Male Masturbaters Professionals?
Male Masturbaters
The 10 Most Infuriating Private ADHD Diagnosis UK Cost
Fails Of All Time Could Have Been Avoided Private Adult Adhd Assessment
10 Things You Learned In Kindergarden That’ll Help You
With SEO Company In London UK saas content marketing company (Jina)
How To Research Anal Plug Online Anal Plug Online – https://Tulun.Ir/User/JosetteF27/ –
The No. 1 Question Everyone Working In Buy Anal Sex Toys Should
Be Able To Answer Anal Toy For Women
How The 10 Most Disastrous Best Panty Vibrator Failures Of All Time Could
Have Been Prevented vibrator under Clothes
Five Killer Quora Answers On Mens Best Sex Toys
mens best sex Toys
20 Insightful Quotes About Automatic Fleshlight multi-Chamber Masturbator
Will Local SEO Marketing Agency Be The Next Supreme Ruler
Of The World? link building Agency uk
The Most Sour Advice We’ve Ever Received On Mesothelioma Lawsuits mesothelioma attorney (http://Www.stes.tyc.edu.tw)
Fridges: It’s Not As Difficult As You Think fridge freezers
uk [https://www.diggerslist.com/6654F5e774f4a/about]
Five Things Everyone Makes Up On The Subject Of
Double Glazed Window Repairs Near Me double glazing repair near Me
You’ll Never Guess This Cost Of Car Key Replacement’s Tricks cost of Car key Replacement
You’ll Never Be Able To Figure Out This
Replacement Volkswagen Keys’s Tricks Replacement volkswagen keys
For Whom Is Replacement Volvo Key And Why You Should Care volvo c30 key replacement
20 Trailblazers Lead The Way In Audi Keys audi connect key
The 10 Most Scariest Things About Audi Car Keys Replacement audi Car Keys replacement
5 Killer Quora Answers To Daftar Situs Togel Daftar Situs Togel
The 10 Most Terrifying Things About Fiat 500 Key Fob Replacement fiat 500 key fob Replacement
Why Nobody Cares About Baby Cot Cribs (Womans-Days.Ru)
10 Unexpected Ferrari Replacement Key Cost Uk Tips Ferrari remote key
Learn What Pragmatic Tricks The Celebs Are Utilizing 프라그마틱 정품확인방법
20 Myths About Private Mental Health Assessment: Debunked Mental Health
Assessment Specialist, Wifidb.Science,
Guide To Gas And Heating Engineer: The Intermediate Guide In Gas
And Heating Engineer Gas And Heating Engineer
You’ll Never Guess This Large Fabric Corner
Sofas’s Benefits fabric two Seater sofa
5 Laws Everybody In Live Casino Should Know 프라그마틱 홈페이지
10 Apps That Can Help You Manage Your Live Casino 프라그마틱 카지노
See What Repair Upvc Windows Tricks The Celebs
Are Utilizing Repair Upvc Windows
Nine Things That Your Parent Taught You About Key Audi key Audi
What Is Handicap Scooters And How To Utilize What Is Handicap Scooters And How To Use electric mobility (Steffen)
10 Tell-Tale Signs You Must See To Find A New Good Accident Lawyers good accident lawyers near me
The Most Profound Problems In Pragmatic Casino 프라그마틱 무료
9 Signs You’re A Double Travel Stroller Expert Pushchairs double
See What Best French Style Fridge Freezer Uk Tricks The Celebs Are Making Use Of
best French style Fridge freezer Uk
5 Laws That Will Help In The Foldable Power Wheelchair Industry electric folding Mobility wheelchair
5 Freestanding Electric Stoves-Related Lessons From The Pros
electric Standing fire (telegra.ph)
7 Easy Tips For Totally Making A Statement With Your Audi Spare Key audi key fob replacement
20 Trailblazers Setting The Standard In Ramp For Wheelchairs walmart Wheelchair Ramps
The 10 Most Scariest Things About Electric Folding Wheelchair Electric folding wheelchair
You’ll Never Guess This Treadmill For Sale’s Tricks Treadmill For sale – https://bookmarkblast.com/,
Audi Key Replacement: 10 Things I’d Like To Have Learned In The Past Audi car key holder
Looking For Inspiration? Check Out Audi A1 Key Audi A5 Key Fob (Squareblogs.Net)
You’ll Be Unable To Guess Built In Microwaves’s Secrets built in microwaves
The 10 Most Terrifying Things About Best Electric Patio Heaters electric
patio heater (Tangela)
How To Identify The Mesothelioma Not Caused By Asbestos That’s Right For You mesothelioma Lawyer
You’ll Never Guess This Power Assisted Self Propelled Wheelchair’s Secrets Power assisted self propelled Wheelchair
Biggest Crypto Casino Tools To Make Your Daily
Life Biggest Crypto Casino Trick That Every Person Must Be Able To biggest crypto casino – Sovren.media,
10 Things Everybody Hates About Programing Key key programmer
What’s The Current Job Market For Mens Masturbation Toys Professionals
Like? mens masturbation toys
20 Things You Should To Ask About Double Glazing Repair Manchester Before You Purchase Double Glazing Repair Manchester window replacement manchester
10 Misconceptions Your Boss Holds About Mystery Box best mystery box sites –
Juanita –
The 10 Most Scariest Things About Legit Crypto Casino legit crypto casino; Milla,
A New York mesothelioma lawyer can assist victims and their families receive
substantial compensation to pay for medical expenses, funeral costs,
travel expenses and more. These attorneys offer a free consultation and streamlined asbestos legal procedure.
How To Recognize The Asbestos Attorney Lawyer Mesothelioma To Be Right For You mesothelioma Lawsuits
The Unknown Benefits Of Mini Replacement Key Fob replacement mini car key, Chelsey,
Modern Freestanding Bioethanol Fireplace Techniques To Simplify Your Daily Lifethe One Modern Freestanding Bioethanol Fireplace Trick Every Person Should Be Able To modern freestanding bioethanol fireplace
Private ADHD Assessment Online Explained In Fewer Than 140
Characters How To Get A Private Adhd Assessment
How To Create An Awesome Instagram Video About ADHD Test
Adult online test for adhd adults (humanlove.stream)
See What Rustic Leather Sofa Tricks The Celebs Are Making
Use Of Rustic leather sofa
Why People Are Talking About Situs Toto Today bandar toto
Nine Things That Your Parent Taught You About Mesothelioma Attorney mesothelioma attorney (elearnportal.science)
5 Must-Know Landrover Keys-Practices You Need To Know For 2023 Land Rover Key
What’s The Job Market For L Shaped Sectional Couch Professionals?
l Shaped sectional couch
Why We Love Couch For Sale (And You Should Also!)
sofa sale
Is Hob The Greatest Thing There Ever Was? Oven
Five Killer Quora Answers On Cot Bed Sales cot bed sales; Deangelo,
Speak “Yes” To These 5 Heaters Electric Fires Tips portable
electric burners (http://Www.google.co.vi)
What’s The Current Job Market For Frost Free Retro
Fridge Freezer Professionals? frost free retro fridge freezer;
Zulma,
Five Killer Quora Answers To Treadmill Electric Treadmill Electric
Responsible For A Glass.Replacement Budget? 12 Tips On How
To Spend Your Money replacement glass for front Door
The Leading Reasons Why People Achieve In The Address
Collection Industry 주소모음 (Lynn)
See What Coolers Wine Tricks The Celebs Are Using coolers Wine
10 Replacement Porsche Key Tricks All Experts Recommend Car key Cutting cost
What’s The Current Job Market For Black Electric Fireplace With
Mantel Professionals? black electric Fireplace with mantel
The Reason Why Everyone Is Talking About Gas Engineers
Near Me Right Now gas heating engineer
A Peek At Online Mystery Box’s Secrets Of Online Mystery Box the best mystery boxes
You’ll Never Be Able To Figure Out This Lpg Gas Engineer Near Me’s Tricks Lpg gas Engineer (http://120.zsluoping.cn/)
Learn To Communicate Retro American Fridge Freezer To Your Boss retro looking Fridge freezer – freebookmarkstore.win,
Two Seater Fabric Sofa Uk Tools To Improve Your Day-To-Day Life
2 seater fabric settee – Alex –
10 Things People Hate About Audi A4 Key Replacement audi A4 replacement key
10 Beautiful Images To Inspire You About Window Glass Replacement Near Me double
Enough Already! 15 Things About Great Crib We’re Tired Of Hearing cribs
Pushchair 2 In 1’s History History Of Pushchair 2 In 1
2 in 1 pushchair
Why Nobody Cares About Treadmills For Home treadmills at home, Siobhan,
20 Important Questions To Be Asking About Buy A Driving License A A1
A2 Without A Test Before You Buy Buy A Driving License
A A1 A2 Without A Test prawo jazdy a1 – Paulette,
10 Audi Keys Meetups You Should Attend Audi keys replacement
10 Tell-Tale Signs You Need To Get A New Link Collection Site 주소모음
9 . What Your Parents Taught You About Electric Fireplace Wall Mounted Electric Fireplace
Wall Mounted (Elden)
See What Replacement Car Key Audi Tricks The Celebs Are
Utilizing Key Audi (Telegra.Ph)
What’s The Job Market For Coffee Machines Capsules Professionals Like?
coffee machines Capsules – coffeee41931.blogdanica.Com –
Patio Pellet Stove Techniques To Simplify Your Daily Lifethe One Patio Pellet Stove Technique Every Person Needs To Know patio Pellet stove
Are The Advances In Technology Making Driving License C+E Better Or Worse?
Ile kosztuje Prawo jazdy na Skuter?
The 9 Things Your Parents Teach You About Togel4d Login Togel4D Login
How Best Cot For Newborn Has Changed The History Of Best Cot
For Newborn best baby cots (Cindy)
Treadmill Incline Foldable Tools To Ease Your Daily Life Treadmill Incline Foldable Trick That
Everybody Should Learn treadmill incline foldable – Kristian –
15 Top Documentaries About Skoda Octavia Replacement Key
Skoda Smart Key
See What Locksmith Near Me For Car Tricks The Celebs Are Using locksmith near me
for car (120.24.213.253)
The 10 Scariest Things About Conservatory Wood Burner conservatory Wood Burner
A Sage Piece Of Advice On Walking Pad Desk From The Age Of Five foldable under desk
treadmill (Chante)
ADD Symptoms In Women Explained In Fewer Than 140 Characters
Adhd And Asd Symptoms
5 Reasons To Be An Online Nissan Key Programming And 5 Reasons Not To Nissan Car Keys
(Telegra.Ph)
You’ll Be Unable To Guess Treadmill Sale UK’s Tricks Treadmill Sale Uk
See What Wall Mounted Fireplace Tricks The Celebs Are Using wall mounted fire
Guide To Emergency Patio Door Repair: The Intermediate Guide Towards Emergency Patio Door Repair Emergency Patio Door Repair
Guide To Walking Pad Standing Desk: The Intermediate Guide The Steps To Walking Pad Standing Desk Walking pad standing Desk
(Git.openprivacy.ca)
How Audi A1 Key Was The Most Talked About Trend In 2023 Audi keyless entry
Bandar Online Togel Tips From The Best In The Industry togel resmi indonesia
How To Tell If You’re In The Right Position To Go After Audi A1
Car Key audi q5 key
You’ll Never Guess This Audi A3 Replacement Key’s Tricks Audi A3 Replacement key
5 Private ADHD Projects That Work For Any Budget uk private adhd assessment (Larry)
5 Killer Quora Answers To Wall Mount Electric Fireplace With
Mantel wall mount electric fireplace with mantel; Randy,
What’s The Current Job Market For Link Togel Professionals Like?
Link togel
What Is The Reason? Coffee Beans Best Is Fast Increasing To Be The Hottest Trend Of 2023 unroasted coffee Beans
It’s True That The Most Common Ultra Lightweight Folding Electric Wheelchair Debate It’s Not As
Black And White As You Might Think lightweight Folding power wheelchair (baidubookmark.com)
The Ultimate Glossary Of Terms About Gas Heater For Outdoor Patio propane gas
patio heater (forum.ressourcerie.fr)
Are You Getting The Most Value The Use Of Your Pragmatic Kr?
프라그마틱 무료체험 슬롯버프 (king-bookmark.stream)
Why No One Cares About Pragmatic Free Slots 프라그마틱 무료 슬롯
Guide To Espresso Machine Sale: The Intermediate Guide Towards Espresso Machine Sale espresso machine Sale
10 Graphics Inspirational About Truck Accident Attorney For Hire
Lawyer Truck Accident
20 Questions You Should Ask About Folding Wheelchair Before You Buy Folding Wheelchair buy folding wheelchair (Hye)
7 Secrets About Online Mystery Boxes That Nobody Will Share With
You mystery box sites (childrenchoir.ru)
10 Tips For Getting The Most Value From Wheelchair Electric Wheelchair electric light weight Wheelchair
(https://mysitesname.com/story7371448/seven-explanations-on-why-electric-wheelchair-uk-is-so-important)
The Often Unknown Benefits Of Tan Leather Chesterfield Tan Leather Chesterfield Sofa (https://Sofas-And-Couches-Uk64308.Fliplife-Wiki.Com)
The Most Sour Advice We’ve Ever Seen About Treadmill For Sale Treadmill For Sale Treadmill Sale
This Is The Ugly The Truth About Buy A Driving License With
Code 95 ile kosztuje prawo jazdy na skuter?
10 Unexpected Crypto Slots Casino Tips casino
crypto games – securityholes.science –
Honda Keys Cut And Program 10 Things I Wish I’d Known In The
Past honda jazz remote key not working; https://troutsailor0.bravejournal.net/searching-for-inspiration,
3 Ways That The Bmw Spare Key Cost Influences Your Life cost Of bmw key replacement
Accident Lawyers Portland 10 Things I’d Loved
To Know Earlier lawyer accident Near me
asbestos settlement was widely used a construction material in the 1930s and into the 1970s.
It was used in pipe insulation and fireproofing, as well as cements, plasters, car breaks, and
much more.
10 Things That Your Family Taught You About Daftar Akun Togel Resmi daftar Akun togel resmi
The 10 Most Scariest Things About Situs Toto Login Situs toto Login
What Is Cars Keys Replacement’ History? History Of Cars Keys Replacement
car keys replacements (Bbs.zhizhuyx.com)
You’ll Be Unable To Guess Togel4d Login’s Tricks togel4d Login
The People Closest To Renault Zoe Key Have Big Secrets To Share renault trafic replacement key – Dianne –
Coffee Machine For Home: It’s Not As Difficult As You Think Coffee machine home
How To Create An Awesome Instagram Video About Car Keys Cut spare car key cutting near
me (Veda)
10 Methods To Build Your Pragmatic Free Slots Empire 프라그마틱 슬롯 체험
The 10 Most Infuriating Link Collection Failures Of All Time
Could Have Been Prevented 링크모음사이트 [Zita]
5 Killer Quora Answers On Toto4d toto4d (Holley)
Where Will Repairing Upvc Windows One Year From What Is
Happening Now? online upvc windows
Why You Should Concentrate On Improving Audi A4 Key Replacement Audi A3 replacement key
Why The Small Wood Burner Is Beneficial When COVID-19
Is In Session wood Stove Burners
Guide To Daftar Akun Togel Resmi: The Intermediate Guide
Towards Daftar Akun Togel Resmi Daftar akun togel resmi
What’s The Current Job Market For Item Upgrades
Professionals Like? item upgrade (Christine)
The Most Hilarious Complaints We’ve Heard About Volvo Key Fobs volvo searching for keys
The Most Convincing Evidence That You Need Asbestos Mesothelioma Compensation mesothelioma attorney
Why Toto Online Terbaik Is So Helpful In COVID-19 bandar Togel Terpercaya
How Much Is A Private ADHD Assessment: What Nobody Is Discussing private adhd assessment lancashire (Norberto)
The People Closest To Treehouse Beds With Slide Uncover Big Secrets boy treehouse bed (https://directmysocial.com/story2743541/why-treehouse-canopy-might-be-your-next-big-obsession)
Guide To 3 Wheel Pushchair Travel System: The Intermediate Guide
To 3 Wheel Pushchair Travel System 3 wheel Pushchair travel system
Best Accident Attorney Tools To Improve Your Daily Life Best Accident Attorney Technique Every Person Needs To Learn accident attorney
9 Things Your Parents Teach You About Wooden Cot Bed Price
wooden cot bed price (http://www.Nzdao.cn)
10 Things That Your Family Taught You About Automatic Folding Mobility
Scooter Uk Automatic Folding Mobility Scooter Uk
10 Best Books On Mesothelioma Attorney mesothelioma claim [lyon-moses-3.technetbloggers.de]
Five Killer Quora Answers On Seat Key Cover seat key cover (Felix)
You’ll Never Guess This Best Accident Lawyer Near
Me’s Secrets best accident lawyer near me
A Look At The Ugly Real Truth Of Driving License Category C Kup Prawo Jazdy A A1 A2 bez testu
See What 2 Seater Chesterfield Couch Tricks The Celebs Are Using 2 seater
chesterfield couch [http://www.youtube.com]
Are You Responsible For An Cost Of Replacement Car Key Budget?
Twelve Top Ways To Spend Your Money Car keys replacement
Five Killer Quora Answers On Gspot Orgasm gspot Orgasm
24-Hours To Improve Adhd Assessment For Adults adhd assessments [Juliann]
An All-Inclusive List Of Lightweight Electric Wheelchairs Folding Uk Dos And Don’ts collapsible Electric Wheelchair Lightweight
Nine Things That Your Parent Taught You About Best SEO Company UK Best seo company uk
Ten Espresso Coffee Makers That Really Make Your Life Better espresso Coffee Makers
15 Great Documentaries About Buying A Mobility Scooter best place to buy mobility scooter (Davida)
You’ll Never Guess This Replacement Window Handle’s Tricks Replacement Window
Trucking Lawyers Near Me Tools To Help You Manage Your
Daily Lifethe One Trucking Lawyers Near Me Trick
That Should Be Used By Everyone Learn trucking lawyers near
me (Annett)
The 3 Greatest Moments In Asbestos Mesothelioma Compensation History mesothelioma lawyers – Hiram –
See What White Wall Mounted Electric Fireplace Tricks The Celebs Are
Using white Wall Mounted electric fireplace
The Three Greatest Moments In Ovens History Best Ovens And Hobs
20 Great Tweets Of All Time Concerning Driving License C+E Ile Kosztuje Prawo Jazdy Na Skuter?
The Best Advice You Could Ever Receive About Key
Car Replacement Cheap Car Key replacement
Folding Treadmill UK Techniques To Simplify Your Everyday Lifethe Only Folding Treadmill UK Technique Every Person Needs To Learn folding Treadmill Uk
5 Bmw 1 Series Key Lessons From Professionals nearby
The 10 Most Scariest Things About Triple Sleeper Bunk Bed triple Sleeper bed
(https://minecraftcommand.science)
What Is The Reason? Replace Window Handle Is Fast Becoming The Hottest Trend Of 2023?
patio door handle Repair near me [https://guerrero-dejesus.technetbloggers.de]
7 Simple Strategies To Completely Moving Your Leather Chesterfield Sofa chesterfield style Corner sofa
9 Signs That You’re The Truck Accident Attorneys Near Me Expert best truck accident lawyer (Krystle)
Need Inspiration? Look Up Treadmill For Sale treadmill For Sale near me
The 15 Things Your Boss Wishes You’d Known About Fabric Corner Couch Modern Fabric Corner Sofa
Ten Things You Learned In Kindergarden Which Will Aid You In Obtaining Mesothelioma Attorney mesothelioma Law firm
9 Lessons Your Parents Teach You About Cot Bed Solid Wood cot bed Solid wood
12 Companies Are Leading The Way In ADHD Adults Symptoms symptoms of adhd
in adults female (http://freeok.Cn/home.php?mod=space&uid=6347118)
15 Great Documentaries About Retro Small Fridge Freezer retro Fridge Freezers best prices
20 Fun Infographics About Gray Fabric Corner Sofa
legend classic back fabric corner sofa – Dustin,
10 Ultra Realistic Sex Doll-Related Ultra Realistic Sex Doll-Related
Projects That Will Stretch Your Creativity realistic Sex doll For Men
9 Things Your Parents Teach You About Boarding Up Commercial Property boarding up commercial property; Valencia,
You’ll Never Be Able To Figure Out This Comfy Couches For Sale’s Tricks comfy couches for sale
The Best Retro Fridge Freezer Uk Tricks To Rewrite Your Life best retro fridge freezer uk – Ahmed,
Undisputed Proof You Need Replacement Audi Key repairing
The 10 Scariest Things About Window Hinge Repair Near Me window Hinge repair
The 3 Greatest Moments In Audi Car Keys History
Replacement Audi A3 key
The 10 Most Scariest Things About Retro Fridge
Freezers For Sale retro Fridge freezers for sale
A Cheat Sheet For The Ultimate On Volvo V50 Key replacement key volvo xc60 (locklear-proctor.Federatedjournals.com)
15 Best Item Upgrade Bloggers You Should Follow Best Item Upgrade
10 Things That Your Family Teach You About Crypto Game Casino Crypto Game Casino
What’s The Current Job Market For Replacement Lexus Key Professionals?
replacement Lexus key
Five Essential Qualities Customers Are Searching For
In Every The Door Doctor window & Door doctor
Some Of The Most Ingenious Things Happening With Buy A
Category B Driving License Without An Exam Kup prawo jazdy z kodem 95 – https://nerdgaming.science/ –
What Auto Locksmiths Near Me Will Be Your Next Big Obsession? auto locksmith key programming near me (Chase)
Guide To 2 Seater Leather And Fabric Sofa: The Intermediate Guide For 2 Seater Leather
And Fabric Sofa 2 Seater Leather And Fabric Sofa – http://Www.Bitsdujour.Com,
10 Tell-Tale Signs You Need To Buy A Double
Car Seat Stroller double buggy newborn – glamorouslengths.com
–
What’s The Current Job Market For Daftar Situs Togel Professionals
Like? daftar Situs togel (togelhongkong62365.blog-kids.com)
Biggest Crypto Casino Tools To Improve Your Daily Life Biggest Crypto Casino Trick Every Individual Should Be Able To biggest Crypto casino
What Is Link Collection Site? What Are The Benefits And
How To Make Use Of It 주소모음사이트
A Complete Guide To Volkswagen Replacement Keys volkswagen car
keys (wise-antelope-hc6hcm.mystrikingly.com)
Guide To Internal Injury Settlement Amounts In 2022 Guide
To Internal Injury Settlement Amounts In 2022 injury attorneys near me
The Best Ovens Uk Tricks To Transform Your Life best ovens
How To Outsmart Your Boss On Mesothelioma And Asbestos Lawyer mesothelioma attorneys
What’s The Current Job Market For Togel Singapore Professionals Like?
togel singapore (Rusty)
What Is Small Lightweight Electric Wheelchair? And How To Use It portable power chair
Why Nobody Cares About Casino Crypto Coin Cryptocurrency Casino
10 Quick Tips For Pragmatic Genuine 프라그마틱 슬롯 (Jeramy)
Why You Should Concentrate On Improving Treadmills For Sale Near Me Smart treadmill
10 Things That Your Family Teach You About Buy A Driving License A A1
A2 Without A Test ile kosztuje prawo jazdy na skuter?
The Intermediate Guide To Upvc Windows Repair repairs to upvc doors
10 Things We All Hate About Window Repair Birmingham Double Glazing Repairs Birmingham Uk
Where Will Private ADHD Assessment Manchester One
Year From In The Near Future? Private ADHD assessment Bristol
cost [Margie]
Five Killer Quora Answers On Bio-Ethanol Fireplace Ethanol
Fireplace (https://138.197.71.160/Fireplacesandstove5888/Electric-Fireplace1119/Wiki/The-Three-Greatest-Moments-In-Freestanding-Fireplace-History)
What Is The Reason? Private ADHD Assessment Near Me Is Fast Becoming The Hottest Trend Of 2023?
private adhd assessments
15 Best Clitoris Vibrator Bloggers You Must Follow Best Clitoris Vibrator
The No. Question That Everyone In Buy A Category B Driving License Without An Exam Needs To Know How To Answer Ile Kosztuje prawo jazdy na skuter?
13 Things About Auto Injury Attorneys You May Not Have Known car injury attorneys Near Me
12 Facts About Buy A Driving License A A1 A2 Without A Test
To Make You Think Smarter About Other People Kup Prawo Jazdy A A1 A2 Bez Testu
How To Save Money On Case Opening Battle case Battle simulator
How To Know If You’re Ready For Seat Key Fob Replacement Seat ibiza key fob not working
Five Things Everyone Makes Up About Case Opening Battles case battle cs (Brayden)
Watch Out: How Virtual Mystery Boxes Is Taking Over And What You
Can Do About It mystery Boxes best
15 Things You’ve Never Known About Sofa Couch For Sale cheap couches For Sale under $200
5 Things That Everyone Doesn’t Know About Scooter Driving License b1 Prawo jazdy
Responsible For The Driving License Category C Budget? 10 Very Bad Ways To Invest Your Money Prawo jazdy C+e
The Most Pervasive Problems In Driving License A1 Kup Prawo Jazdy Kategorii B Bez Egzaminu
Way cool! Some very valid points! I appreciate you
penning this post plus the rest of the site is very good.
Hey There. I found your blog using msn. This
is an extremely well written article. I will be sure to bookmark it and return to read more of your useful info.
Thanks for the post. I will definitely return.
Hello, i read your blog from time to time and i
own a similar one and i was just curious if you get a lot
of spam remarks? If so how do you reduce it, any plugin or anything you can recommend?
I get so much lately it’s driving me crazy so any
assistance is very much appreciated.
Do you mind if I quote a couple of your posts as long
as I provide credit and sources back to your blog?
My blog is in the very same niche as yours and my users would
really benefit from a lot of the information you provide here.
Please let me know if this alright with you. Thanks!
Burlesque Show OP; Oplovesite.com,
What’s up, after reading this remarkable article i am also cheerful to
share my know-how here with mates.
We’re a group of volunteers and opening a new scheme in our
community. Your web site provided us with valuable info to work on. You have done a formidable job and our entire community will be grateful to you.
Greetings! Very useful advice in this particular article!
It is the little changes which will make the largest changes.
Thanks for sharing!
Hi there, You have done a fantastic job. I’ll
certainly digg it and personally recommend to my friends.
I am sure they’ll be benefited from this website.
Wow that was odd. I just wrote an extremely long comment but after I clicked submit my comment didn’t show up.
Grrrr… well I’m not writing all that over again. Anyway, just wanted to say wonderful blog!
What’s up mates, how is everything, and what you
would like to say regarding this post, in my view its actually
remarkable designed for me.
Great article! This is the type of information that are meant to be shared
across the internet. Disgrace on Google for not positioning this put up higher!
Come on over and talk over with my website . Thanks =)
Thank you for the good writeup. It in fact was a amusement
account it. Look advanced to far added agreeable from you!
However, how could we communicate?
Hi there, i read your blog occasionally and i own a similar one and i was just curious if you get
a lot of spam responses? If so how do you protect against it,
any plugin or anything you can advise? I get so much lately it’s driving
me crazy so any help is very much appreciated.
Appreciate this post. Will try it out.
I’ve read several just right stuff here. Definitely worth
bookmarking for revisiting. I surprise how much effort you place to make this type
of wonderful informative site.
Thanks for the auspicious writeup. It actually was once a entertainment account it.
Glance advanced to more added agreeable from you!
However, how could we keep up a correspondence?
Hello, just wanted to tell you, I liked this article.
It was practical. Keep on posting!
Hey! Would you mind if I share your blog with my myspace group?
There’s a lot of folks that I think would really appreciate your content.
Please let me know. Thank you
What’s up, its fastidious paragraph regarding media print,
we all know media is a impressive source of information.
you’re in point of fact a good webmaster. The website loading pace is amazing.
It kind of feels that you’re doing any distinctive trick.
In addition, The contents are masterpiece. you’ve done
a excellent process in this topic!
Thank you for the good writeup. It in fact was a amusement account it.
Look advanced to more added agreeable from you!
However, how can we communicate?
I really like what you guys tend to be up too.
This sort of clever work and reporting! Keep up the great works guys I’ve added you guys to my own blogroll.
Hi, I do think this is a great blog. I stumbledupon it 😉 I am going to come back once
again since i have saved as a favorite it.
Money and freedom is the greatest way to change, may you be rich and continue
to help others.
certainly like your web site but you need to check the
spelling on several of your posts. A number of them are rife with spelling issues and I
to find it very bothersome to inform the reality on the other hand I
will certainly come back again.
I’ve been surfing online more than 3 hours today, yet I never found any interesting article like yours.
It’s pretty worth enough for me. In my view, if all webmasters and bloggers made good content
as you did, the internet will be much more useful than ever
before.
I’m really impressed together with your writing abilities
as neatly as with the format to your weblog. Is that this a
paid theme or did you modify it yourself? Anyway keep up the nice quality
writing, it is rare to see a great blog like this one nowadays..
I’m not that much of a online reader to be honest but your sites really nice, keep it up!
I’ll go ahead and bookmark your website to come back later on. All
the best
Spot on with this write-up, I seriously believe this website needs a lot more attention. I’ll probably be
returning to read more, thanks for the information!
Saved as a favorite, I really like your website!
Its such as you learn my thoughts! You appear to understand
a lot approximately this, such as you wrote the
e book in it or something. I feel that you could do with some p.c.
to pressure the message home a bit, however instead of that, that
is great blog. An excellent read. I will definitely be back.
I was recommended this blog by way of my cousin. I am now not sure whether this submit is written through
him as no one else realize such special approximately my difficulty.
You are incredible! Thank you!
I used to be recommended this blog through my cousin. I’m no longer certain whether this
put up is written by way of him as no one else know such detailed approximately
my problem. You are incredible! Thanks!
There’s certainly a lot to learn about this subject.
I really like all the points you’ve made.
Hi there, I found your website by means of Google whilst looking for a related matter,
your web site got here up, it seems great. I have
bookmarked it in my google bookmarks.
Hi there, simply was aware of your weblog thru Google, and located that it’s really informative.
I’m gonna watch out for brussels. I’ll be grateful in case you proceed this in future.
A lot of other people will likely be benefited out of your writing.
Cheers!
If you are going for finest contents like I
do, just pay a visit this web site daily as it provides feature contents, thanks
Hey there, You’ve done a fantastic job. I’ll definitely digg it and personally
recommend to my friends. I’m sure they’ll be benefited from this website.
I always used to study post in news papers but now as I am a user of
internet so from now I am using net for articles,
thanks to web.
If you desire to get much from this article then you have to apply
such techniques to your won web site.
We stumbled over here by a different page and thought I
might as well check things out. I like what I see so now i’m following
you. Look forward to going over your web page for a second time.
Hi there, just became alert to your blog through Google, and
found that it’s really informative. I am going
to watch out for brussels. I will be grateful if you continue this in future.
A lot of people will be benefited from your writing.
Cheers!
Incredible! This blog looks just like my old one! It’s on a completely different topic but it has
pretty much the same page layout and design. Wonderful choice of colors!
I need to to thank you for this wonderful
read!! I absolutely enjoyed every bit of it.
I’ve got you bookmarked to check out new stuff you post…
Howdy I am so glad I found your web site, I really found
you by accident, while I was browsing on Digg for something
else, Anyways I am here now and would just like to say thanks a lot for a incredible post and a all
round thrilling blog (I also love the theme/design),
I don’t have time to browse it all at the moment but I have bookmarked it and also included your RSS feeds, so
when I have time I will be back to read more, Please do keep up the superb work.
Great delivery. Great arguments. Keep up the good effort.
Great website. Plenty of helpful info here. I’m sending it to several friends ans additionally sharing in delicious.
And certainly, thank you on your effort!
This information is worth everyone’s attention. When can I find out more?
The Evolution Of Local SEO London seo east London
Guide To Accident Claim Lawyers: The Intermediate Guide To Accident Claim Lawyers accident claim Lawyer
Guide To Double Glazing Doctors: The Intermediate Guide
For Double Glazing Doctors double glazing Doctors
5 Killer Quora Answers To Leather 4 Seater Recliner
Sofa Leather 4 seater recliner Sofa
The 10 Most Dismal Head Injury Settlement Amount Fails Of All Time Could Have Been Prevented lawyer near me injury (telegra.ph)
What Is Cryptoslots Casino And How To Use It Casino Crypto
15 Best Robot Vacuum UK Bloggers You Need
To Follow Best robot vacuum uk (Astrid)
You Are Responsible For A Asbestos Mesothelioma Lawsuit Budget?
12 Top Ways To Spend Your Money asbestos lawsuit (Virginia)
Five People You Should Know In The Truck Accident Attorneys Industry Big truck accident attorney
How To Find The Perfect Train Accident Claim On The Internet semi truck accident lawyers
How To Create An Awesome Instagram Video About
Tilt And Turn Window Mechanism Tilt and turn window mechanism broken (sciencewiki.science)
One Of The Most Innovative Things Happening With Asbestos Lawsuit Asbestos Lawsuits
What Freud Can Teach Us About Train Accident Lawyer commercial truck
accident attorneys – Micki –
20 Up-Andcomers To Watch The How To Get ADHD Medication Uk Industry Inattentive Adhd Medication Adults
See What Auto Key Repair Near Me Tricks The Celebs Are Using Key repair near me
The 10 Most Terrifying Things About Robot Vacuum Cleaner Robot Vacuum Cleaner
Uk (Daimler-Bkk.Portal-Gesundheitonline.De)
14 Businesses Doing An Amazing Job At Getting A Car Key Cut key cut service
The Companies That Are The Least Well-Known To Keep An Eye
On In The Minnesota Birth Injury Attorneys Industry lawyer near Me injury
8 Tips To Increase Your Link Collection Game 링크모음사이트
What’s The Current Job Market For Private ADHD Diagnosis
UK Professionals? private Adhd diagnosis uk (http://www.hondacityclub.com/all_new/home.php?mod=space&uid=1726678)
A Comprehensive Guide To ADHD Private Diagnosis Cost. Ultimate Guide
To ADHD Private Diagnosis Cost private adhd Assessment cost
Who Is Head Injury Claim Compensation And Why You Should Care best injury
lawyer near me (Christiane)
You’ll Never Guess This Best Car Locksmith Near Bedfordshire’s Tricks best car locksmith Near bedfordshire
The Leading Reasons Why People Perform Well In The Case Opening Battle
Industry cs Battle Case
How To Determine If You’re In The Right Place To
Best Affordable Coffee Machine Best cappuccino Machine (minecraftcommand.science)
You’ll Never Guess This Best Accident Lawyer Near Me’s Secrets Best
Accident Lawyer Near Me (Click4R.Com)
The 10 Most Popular Pinterest Profiles To Keep Track Of Online Crypto Casino Best online crypto casino
9 Things Your Parents Taught You About Upvc Door Panel
Inserts Upvc Door Panel Insert (Lovewiki.Faith)
What Is The Reason Auto Accident Lawsuits Is Right For You accident car lawyer
Are You Sick Of CSGO Case Battle? 10 Inspirational Sources That Will Invigorate Your Love case battles (Ara)
20 Misconceptions About Asbestos Cancer Lawyer Mesothelioma Settlement:
Busted Mesothelioma Attorneys
Ten Ways To Build Your Window Repair Luton Empire double glazing repairs
near me – https://maps.google.com.sl/ –
Five Commercial Truck Accident Lawyer Lessons From The Professionals Best Semi Truck Accident Attorney
A Time-Travelling Journey What People Said About Best Bunk 20 Years Ago
Adult Bunkbed
The 10 Most Terrifying Things About Item Upgrade item upgrade (bookmarking.win)
14 Businesses Doing A Great Job At Electric Fire Suites Oak electric fireplace suites near me (http://www.scdmtj.com)
10 Misconceptions Your Boss Holds Regarding Leather Couch 4 Seater four seater leather lounge
11 Strategies To Completely Block Your Hinges double glazed window hinge repair (Erin)
10 Things Everybody Hates About Asbestos Mesothelioma Lawyers mesothelioma lawsuit
What’s The Job Market For Large Wood Burning Stove Professionals?
large wood burning stove
The Most Common Auto Lawyers Near Me Mistake Every Beginner Makes attorney near me car accident (Margie)
Five Killer Quora Answers On Reconditioned Dual Fuel Range
Cookers reconditioned dual fuel range cookers
Learn What Asbestos Attorney Lawyer Mesothelioma Tricks The Celebs Are Making Use Of mesothelioma lawsuit
10 Misconceptions Your Boss Holds Regarding Item Upgrade item upgrader kit (Mariel)
5 Killer Quora Answers To Hob Uk hob uk [Pedro]
3 Common Causes For Why Your Car Accident Attorney
Isn’t Working (And How To Fix It) Car accident attorney near me
20 Questions You Must Always ASK ABOUT Westfield Birth Injury
Attorneys Before Buying It Injury lawyers
Are You Responsible For The Virtual Mystery Boxes Budget?
10 Amazing Ways To Spend Your Money online mystery box opening
4 Dirty Little Details About The Pragmatic Sugar
Rush Industry 프라그마틱 정품 확인법, https://www.google.com.co/Url?q=https://writeablog.net/carwasher7/the-reasons-pragmatic-is-everywhere-This-year,
20 Up And Coming Mystery Box Stars To Watch The Mystery Box Industry best mystery box online shop
Buzzwords De-Buzzed: 10 Alternative Methods To Say Automatic Vacuum And Mop Best Automatic Vacuum
10 Things Everybody Hates About Nissan Car Key Programmers
20 Best White Bunk Beds Websites Taking The Internet By Storm
Best Place To Buy Bunk Beds Near Me (Vuf.Minagricultura.Gov.Co)
9 Lessons Your Parents Taught You About Accident Attorneys Near Me Accident attorneys near Me (timeoftheworld.Date)
11 Methods To Totally Defeat Your ADHD Women Test Add women
20 Myths About Mesothelioma And Asbestos Lawyer: Busted asbestos lawyers [Tracey]
This History Behind Skoda Car Key Replacement Will Haunt You Forever!
Skoda Octavia 2 Key Programming
Five Essential Tools Everyone Is In The Washington Birth Injury Attorneys Industry Should Be
Making Use Of injurys attorney near Me
Are You Responsible For An Accident And Injury Attorneys Budget?
12 Ways To Spend Your Money Accident And Injury Lawyers
11 Creative Ways To Write About Collapsible Scooters boot
mobility scooter; https://saveyoursite.date/story.php?title=question-how-much-do-you-know-about-Boot-mobility-scooters,
Where Is Drip Coffee Be One Year From What Is Happening Now?
dripping coffee machine
Guide To Replacement Sash Windows: The Intermediate Guide In Replacement Sash Windows Replacement Sash windows
15 Gifts For The Car Attorneys Near Me Lover In Your Life car injury attorney Near
me (http://www.pocock.com)
10 Wrong Answers To Common Truck Accident Attorneys Questions: Do You Know The Right Ones?
big truck accident lawyer
Guide To Accident Claim Lawyers: The Intermediate Guide For Accident Claim Lawyers accident Claim lawyer
Guide To Outside Wood Burners: The Intermediate Guide
To Outside Wood Burners Outside Wood Burners – http://Www.Metooo.Es,
Where Can You Find The Top Best Leather Sofa Information? leather couch (Aurora)
The Main Issue With Hire Auto Accident Attorney, And How You Can Fix It Car
Accident Injury Attorneys (Prpack.Ru)
10 Apps That Can Help You Manage Your Address Collection 링크모음
What Is ADHD Treatment For Adults? History Of ADHD Treatment For Adults what is adhd treatment for adults [Michelle]
10 Things Your Competitors Can Teach You About Upvc Doors Repair upvc doors repairs near me
What Is The Future Of Renault Captur Key Be Like In 100 Years?
renault clio locked keys in Car
Double Glazed Window Installers Tools To Make Your Daily Lifethe
One Double Glazed Window Installers Trick Every Person Should Know
Double Glazed Window Installers – https://Nerdgaming.Science/Wiki/10_Double_Glazing_Window_Installation_Related_Projects_That_Can_Stretch_Your_Creativity –
Sectionals For Sale Near Me Tools To Streamline Your Daily Life Sectionals For Sale Near
Me Technique Every Person Needs To Know Sectionals For Sale Near Me
Speak “Yes” To These 5 Bunk Beds For Adults Tips bunk beds Children’s
10 Meetups About Mesothelioma Asbestos Claims You
Should Attend mesothelioma lawsuit (http://www.metooo.io)
The Next Big Event In The Best Robot Vacuum That
Mops Industry automatic vacuum Cleaner
Five Killer Quora Answers To Dual Fuel Range
Cooker dual fuel range cooker
What Is The Heck What Exactly Is Door Doctor Near Me?
Window Sealing
10 Places Where You Can Find Wall Mounted Ethanol Fireplace wall mounted bioethanol fire
The Not So Well-Known Benefits Of Small Wood Burner Very Small Wood Burner
5 Cabin Beds Leçons From The Pros cabin bed with sofa underneath (https://jantzen-elgaard-2.blogbright.net/10-cabins-bed-related-meetups-you-should-attend)
20 Fun Details About Emergency Locksmith Nearest Locksmith
9 Lessons Your Parents Teach You About Leather Pull Out Couch leather pull Out couch
What Do You Do To Know If You’re Ready To Go After Link Collection 주소모음사이트 [Bonny]
9 Lessons Your Parents Teach You About Best Car Locksmiths In Milton Keynes
car locksmiths in milton keynes
Adult Toy Store Near Me: The Good, The Bad, And The Ugly adult toys for sale near me; http://bstm.kr/bbs/board.php?bo_table=free&wr_id=667939,
What Experts In The Field Would Like You To Be Able To how to start jaguar xf without smart key
8 Tips To Improve Your Spare Key For Cars Game car remote key
20 Resources To Make You Better At Ramps For Wheelchairs wheelchair threshold ramp, Maisie,
5 Laws Anybody Working In Jaguar Xf Key Should Know Broken car Key
The 10 Scariest Things About Case Opening Battle Case opening battle
Midi Bed With Storage 10 Things I Wish I’d Known In The Past pine mid sleeper cabin bed
Five Killer Quora Answers On Upvc Door Infill Panel upvc Door Infill Panel
Wow! At last I got a weblog from where I know how to in fact obtain valuable
data regarding my study and knowledge.
Will Case Battle Ever Be The King Of The World?
Case Battle Win – http://Www.Youtube.Com,
10 No-Fuss Strategies To Figuring Out Your
Milton Keynes Double Glazing Glass cutting milton keynes – olderworkers.com.au –
How The 10 Most Disastrous Pragmatic Sugar Rush Failures Of
All Time Could Have Been Prevented 프라그마틱 정품확인방법
The Top Folding Mobility Scooter For 25 Stone The Gurus Are Using Three Things cheap foldable mobility Scooter
10 Facts About Keys For Hyundai Cars That Will Instantly Get You Into A Great Mood Keys Hyundai
15 Documentaries That Are Best About Wall Mount Fireplace fireplace insert, Raumlaborlaw.Com,
Why 1kg Coffee Beans Price Is Your Next Big Obsession? 1kg of coffee beans
See What Lightweight Motorized Folding Wheelchair
Tricks The Celebs Are Making Use Of Lightweight Motorized Folding Wheelchair
This Story Behind Replacing Volkswagen Key Will Haunt You For The
Rest Of Your Life! Programming
Attorneys Accidents Tools To Ease Your Everyday Lifethe Only Attorneys Accidents Trick Every Person Should Learn Attorneys Accidents
5 Killer Quora Answers To Wood Burning Stoves Uk wood burning stoves uk (http://79bo.com/)
The 10 Scariest Things About Best Accident Attorney Near Me accident attorney near
me, squareblogs.net,
Guide To Patio Sliding Doors Repair: The Intermediate Guide For Patio Sliding Doors Repair
patio sliding doors repair – Domingo –
The Best Car Accident Lawyers Near Me Tricks To Transform Your
Life Best car accident lawyers Near me (smzpp.com)
5 Killer Quora Answers On Mesothelioma Asbestos Lawyers
Mesothelioma Attorney
The 10 Scariest Things About Faux Leather Couch Faux Leather Couch
Heya i am for the first time here. I found this board and I find It
really useful & it helped me out much. I hope to give something back and aid others like you aided
me.
What Is Kia Key Replacement? History Of Kia Key Replacement kia carens key
What Is Auto Accident Lawyer And Why Is Everyone Speakin’
About It? car crash Attorneys near me
5 Lessons You Can Learn From Lost Ferrari Key ferrari key 2018 (Marcos)
Aw, this was an extremely good post. Finding the time and actual effort to produce a superb article… but what can I say…
I put things off a lot and never seem to get anything done.
9 Things Your Parents Taught You About Amazon Patio Heater Electric
amazon patio heater electric – Floyd –
How To Explain Best Truck Accident Attorney To Your Grandparents Attorneys For Truckers
How Hinges Was The Most Talked About Trend In 2024 how to replace a hinge on a double glazed window
Do you mind if I quote a few of your articles as long as I provide credit and sources
back to your blog? My blog site is in the exact same
niche as yours and my users would truly benefit from some of the
information you present here. Please let me know if this
ok with you. Cheers!
The Unspoken Secrets Of Patio Door Repair tilt and
slide patio door repairs (https://enevoldsen-hubbard-2.hubstack.net/watch-out-what-patio-sliding-doors-repair-is-taking-Over-and-what-you-can-do-about-it)
Why Is Blue Leather Chesterfield So Popular?
blue leather chesterfield sofa (Stefan)
I know this web site presents quality depending articles or reviews and other material, is
there any other website which offers such things in quality?
The Ultimate Glossary Of Terms About Window Handles Replacement Window handles for upvc
Where Can You Find The Most Reliable Double Glazing Repairs
Manchester Information? upvc
8 Tips To Improve Your Portable Mobility Scooters For Sale Game mobility scooter near me for Sale
What’s The Job Market For 4 Seat Leather Sofa Professionals?
4 seat Leather sofa (Telegra.ph)
Item Upgrading: 11 Thing You’re Leaving Out upgrade item (Janeen)
7 Simple Tricks To Totally Cannabis-Infused Pod Coffee
Machines Ground coffee machines
How Can A Weekly Black Friday Power Tool Deals Project Can Change Your Life online Tools Store
See What Trucking Accident Lawyer Near Me Tricks The Celebs Are Making Use Of Trucking Accident Lawyer Near Me
Ten Things You Learned About Kindergarden That Will
Aid You In Obtaining Car Accident Attorney
For Hire car wreck attorneys near me
10 Unexpected Auto Accident Tips car collision lawyers near Me
I’m really enjoying the design and layout of your blog.
It’s a very easy on the eyes which makes it much more enjoyable for me to come here and visit more often. Did you hire out a
developer to create your theme? Great work!
The 12 Best Comfy Couch UK Accounts To Follow On Twitter couches on sale near me;
Mervin,
10 Myths Your Boss Is Spreading About Best Accident Lawyer Near Me injury
The Ugly The Truth About Hire Car Accident Attorneys Car Wreck Lawyers Near Me
Meet Your Fellow Nissan Key Fobs Enthusiasts. Steve Jobs Of The Nissan Key Fobs Industry car Key replacement nissan
The Reason You Shouldn’t Think About The Need To Improve Your Mystery Box mystery Box website
The People Nearest To Locksmith For Cars Have
Big Secrets To Share nearest
3 Ways In Which The Car Locksmiths Near Bedfordshire Influences Your Life car locksmith near bedfordshire
5 Injury Compensation Projects For Any Budget Good Injury
Lawyers Near Me [Telegra.Ph]
What’s The Job Market For Conservatory Door Repairs Professionals?
conservatory door Repair
You’ll Be Unable To Guess Trucking Lawyers’s Tricks
trucking lawyers [https://king-wifi.win/wiki/5_reasons_to_be_an_online_truck_accident_attorney_near_me_shop_And_5_reasons_to_not]
10 Tell-Tale Warning Signs You Need To Know Before You Buy Mesothelioma Asbestos Lawyers mesothelioma Lawyers (http://taikwu.com.tw)
The Reason Why Everyone Is Talking About Pragmatic Ranking Right Now 프라그마틱 카지노
What The 10 Most Worst Car Remote Key Repair FAILURES Of
All Time Could Have Been Prevented Car Remote Key Repair Near Me
(Hefeiyechang.Com)
What Asbestos Cancer Law Lawyer Mesothelioma Settlement Experts Want You To Learn Mesothelioma attorneys
10 Things That Your Family Teach You About Locksmith Near Me For Cheap Locksmith Near Me For Cheap
9 . What Your Parents Taught You About Best Gas Patio Heater Uk
gas patio heater uk
Five Twin Tree House Bunk Bed Lessons From Professionals bunk bed treehouse (bookmarkfox.com)
10 Things You Learned In Preschool That Can Help You In Asbestos Mesothelioma
Compensation mesothelioma attorney (https://Www.Thehomeautomationhub.com)
What’s The Current Job Market For Robot Vacuum With Mop Professionals Like?
Robot vacuum with mop
See What Baby Cot Bed Tricks The Celebs Are Using Baby Cot Bed
Asbestos Cancer Lawsuit Lawyer Mesothelioma Settlement: What Nobody Is Discussing asbestos lawyers (https://elearnportal.science/wiki/The_Top_Reasons_Why_People_Succeed_At_The_Asbestos_Cancer_Law_Lawyer_Mesothelioma_Settlement_Industry)
“Ask Me Anything:10 Responses To Your Questions About Mesothelioma And Asbestos Lawyer Asbestos lawyers (morphomics.science)
What Is Auto Accident Injury Attorney? What Are The Benefits And How To Make Use Of
It lawyers for car accident near me
Seven Explanations On Why Misted Glass Repair Is Important can you repair misted double glazed windows
What’s The Current Job Market For Power Tools Shops Near Me Professionals?
tools Shops Near Me (Fatahal.com)
7 Little Changes That Will Make The Difference With Your ADHD Testing private adhd tests
– Frank –
Watch Out: What Sliding Sash Window Is Taking Over And What You Can Do
About It Box Sash Windows
What’s The Current Job Market For Accident Lawyer Near Me
Professionals Like? Accident Lawyer near Me
9 Lessons Your Parents Taught You About Upvc Door With Side Panel upvc door with side panel
Buggy Single Tools To Streamline Your Daily Life Buggy Single Technique Every
Person Needs To Know Buggy single
Learn To Communicate Mystery Box To Your Boss mystery boxes (https://www.youtube.com/redirect?q=https://posteezy.com/5-killer-quora-answers-mystery-boxes-0)
7 Simple Tips To Totally Rocking Your Asbestos Lawsuit Attorneys Mesothelioma Lawyer (47.108.249.16)
The 10 Most Scariest Things About ADHD Hyperactivity Symptoms
In Adults adhd hyperactivity symptoms in adults (k12.instructure.com)
Right here is the perfect blog for anybody who would like
to find out about this topic. You know so much its almost tough to argue with you (not that I actually will need to…HaHa).
You certainly put a brand new spin on a topic that’s been written about for a long
time. Great stuff, just excellent!
Guide To Robot Vacuums Best: The Intermediate Guide Towards
Robot Vacuums Best robot vacuums best
What’s The Job Market For Accident Injury Attorneys Near Me Professionals Like?
accident injury attorneys near me
10 Glass Repairs Windows Tips All Experts Recommend Glass Door lock repair
See What Two Seater Fabric Sofa Uk Tricks The Celebs Are Using two seater fabric sofa (Shellie)
It’s The Best Affordable Robot Vacuum Case Study You’ll
Never Forget Best Auto Vacuum
5 Killer Quora Answers On Composite Door Glass Replacement composite Door glass replacement
Are You In Search Of Inspiration? Check Out Power Tools Uk power tools
You’ll Be Unable To Guess Attorney For Birth Injury’s Secrets injury – Epifania,
3 Ways The Car Lawyers Near Me Can Affect Your Life accident Car attorney
Door And Window Doctor Tools To Help You Manage Your Daily Lifethe One Door And Window Doctor
Trick That Should Be Used By Everyone Learn door and window doctor – Notabug.org –
15 Terms That Everyone Is In The Door Repair Near Me Industry Should Know window replacement near Me
Five Killer Quora Answers To Accident Attorney Near Me accident attorney Near me
How To Make An Amazing Instagram Video About Locksmith For
Cars car Auto locksmith near me
Your Family Will Thank You For Getting This Where To Get Honda Key Cut replacement Honda key
This Week’s Most Remarkable Stories About Wall-Mounted Fireplace Electric Fireplaces (losprinters.ru)
20 Inspiring Quotes About Replacement Windows
Birmingham double glazing company birmingham (Spencer)
Accident Injury Attorney Techniques To Simplify Your
Daily Life Accident Injury Attorney Technique Every Person Needs To Know accident Injury attorney – scientific-Programs.science,
The Story Behind Traffic Accident Lawyer Near Me Will Haunt
You Forever! injurys attorney near me
Why Asbestos Attorney Lawyer Mesothelioma Should Be Your Next Big Obsession? Mesothelioma Attorneys
10 Healthy Accident Claim Lawyers Habits Lawyers for injurys near me
How To Determine If You’re Set To Go After Squirtingdildos strapon That Squirts
10 Things We All Hate About Robot Vacuum
Cleaner robot vacuum cleaners Best
4 Dirty Little Tips About The Pragmatic Slot Recommendations Industry 프라그마틱 무료게임
15 Bizarre Hobbies That’ll Make You More Successful At Address Collection Site 주소모음 (http://Www.Cksschool.com)
Admiring the time and effort you put into your website and
detailed information you offer. It’s awesome to come across a blog
every once in a while that isn’t the same outdated rehashed material.
Great read! I’ve bookmarked your site and I’m including your RSS feeds to my Google account.
Are You In Search Of Inspiration? Check Out Replacement Windows Near Me windows Replacement; wayranks.com,
The 10 Worst Outdoor Wood Burner Fails Of All Time Could Have Been Prevented small wood burner for shed uk [Mose]
You’ll Never Be Able To Figure Out This Misted Double Glazing Repairs Near Me’s Benefits misted double
glazing (http://srv29897.ht-test.Ru)
11 “Faux Pas” That Are Actually OK To Create Using Your Hob oven hob (Bertha)
30 Inspirational Quotes For Mens Masturbator Best Masterbater
You’ll Never Guess This Car Key Replacement Near
Me’s Benefits car key replacement near me – Nicolas,
I’m not sure why but this blog is loading
extremely slow for me. Is anyone else having this problem or is it a issue on my end?
I’ll check back later and see if the problem still exists.
Nine Things That Your Parent Taught You About Glass
Repair Manchester Glass Repair Manchester
(Whorlmodem27.Bravejournal.Net)
15 Inspiring Facts About Replacement Key Bmw You Didn’t Know Bmw keys not working
15 Up-And-Coming Media Wall Electric Fireplace Bloggers You Need To
Follow Inset media wall Fire
A Look Into The Secrets Of Locksmith Cars Keysmith Car Near Me
Why Pragmatic Is Your Next Big Obsession? 프라그마틱 슬롯
The Best Place To Research Online Power Tools Online
power tools Online
The Most Innovative Things Happening With Best Car Accident Attorney best attorney for
car accident – https://cameradb.review/wiki/7_Simple_Changes_Thatll_Make_A_Big_Difference_With_Your_Car_Attorney_Near_Me,
3 In 1 Rollator Walker Techniques To Simplify Your Daily Life
3 In 1 Rollator Walker Trick Every Individual Should Know 3 In 1 Rollator
How To Tell If You’re In The Right Place To Single Oven Fan oven buying guide
Car Accident Attorneys: What Nobody Is Talking About Car accidents attorney
Hi mates, pleasant paragraph and fastidious urging commented at
this place, I am genuinely enjoying by these.
How To Explain Window Hinge Repairs Near Me To Your Grandparents wood window Repairs near me
15 Strange Hobbies That Will Make You Smarter
At Lawyers For Accidents Near Me Accident Injury law firm
10 Meetups About Test For Adult ADHD You Should Attend How
do you test for adhd in adults – http://eric1819.com/home.php?mod=Space&uid=828063,
The 10 Scariest Things About Trucking Accidents Attorneys Trucking accidents Attorneys
The Next Big Trend In The Jaguar Xf Key Fob Replacement Industry jaguar xe Keyless Entry not working
20 Myths About Replace Lock On Upvc Door: Debunked Replacement Window Locks
All The Details Of Class Action Lawsuit Asbestos Exposure Dos And Don’ts asbestos Lawsuits (zenwriting.net)
15 Terms That Everyone Involved In Asbestos Cancer Lawyer Mesothelioma Settlement Industry Should Know Mesothelioma Attorney
What’s The Reason You’re Failing At Baltimore Accident Lawyers Accident lawsuits
The Unknown Benefits Of Boarding Up Windows Boarding Up Windows Service
20 Insightful Quotes About Lawyer For Car Accidents car accident attorneys near me
9 Things Your Parents Taught You About Upvc Window Repair Near Me window repair near me
The Reason Behind Aluminium Window Handle Is The Most Popular Topic In 2024 window handle
replacements and repairs (Tayla)
10 Ways To Build Your Car Accident Lawsuit Empire car accident lawyer
best; Loyd,
Its like you read my mind! You appear to know so much about this,
like you wrote the book in it or something.
I think that you could do with some pics to drive the message home a bit, but other than that, this is fantastic blog.
A great read. I will certainly be back.
Is Sex Toys For Him The Best Thing There Ever
Was? adult male toys; Dominic,
How A Weekly Replacing Upvc Window Handles Project Can Change Your Life patio door handle repair near me,
Laurinda,
See What Wooden Sash Windows Tricks The Celebs Are Using wooden sash windows
Five Killer Quora Answers To Lawyers For Accidents Near Me Lawyers For Accidents Near Me
The Houston Asbestos Attorney Success Story
You’ll Never Be Able To mesothelioma lawyers
10 Top Facebook Pages Of All Time About Back Injury Compensation injury claim lawyer
I loved as much as you’ll receive carried out right here.
The sketch is tasteful, your authored subject matter stylish.
nonetheless, you command get bought an nervousness over
that you wish be delivering the following. unwell unquestionably come more
formerly again since exactly the same nearly a lot often inside case you shield this hike.
You’ll Be Unable To Guess Upvc Windows Manchester’s
Secrets pvc windows manchester
Quiz: How Much Do You Know About Semi Truck Accident Attorney?
lawyers for truck Drivers
What’s The Current Job Market For Mercedes Replacement Key Cost Uk
Professionals Like? mercedes replacement key
Guide To Trucking Lawyers: The Intermediate Guide For Trucking
Lawyers trucking lawyers
9 Lessons Your Parents Teach You About Couples Sex Machines sex Machines
Hi to every body, it’s my first pay a quick visit of this website; this web site contains amazing and genuinely good
data designed for visitors.
Hi, I do think this is an excellent web site. I stumbledupon it 😉 I am going to return yet again since i have
saved as a favorite it. Money and freedom is the best way
to change, may you be rich and continue to guide
others.
5 Laws That Will Help The Misted Sealed Units Industry Misted Windows
What’s The Current Job Market For Accident &
Injury Lawyers Professionals? Accident & Injury Lawyers
15 Up-And-Coming Emergency Locksmith Service Near Me Bloggers You
Need To See local locksmith service (Rosaura)
5 Must-Know Auto Accidents Lawyers Techniques To Know For 2023 Car Injury Lawyers Near Me
12 Companies Are Leading The Way In Auto Lawyers car injury attorney near me
(Kandice)
What Is Truck Accident Lawyer Near Me And Why Is Everyone Speakin’ About It?
truck Crash attorney
15 Gifts For The Truck Accident Attorneys Lover In Your Life best semi truck Accident attorney
8 Tips For Boosting Your Auto Accident Attorney For Hire Game car accident lawyers no injury
Be On The Lookout For: How Hire Truck Accident Lawyer Is Taking Over The World And What We Can Do
About It commercial truck accident Lawyers
The Intermediate Guide For Car Accident Attorneys car accident Injury lawyers
All Natural Homemade Acne Soap HiOP
Here’s An Interesting Fact Regarding Truck Lawyers Near Me truck accidents Lawyer (http://www.deepzone.Net)
Hey! I know this is somewhat off-topic but I needed to ask.
Does operating a well-established blog like yours require a lot of work?
I’m completely new to blogging but I do write in my journal daily.
I’d like to start a blog so I can easily share my own experience and thoughts online.
Please let me know if you have any kind of suggestions or tips for brand new aspiring bloggers.
Appreciate it!
It Is Also A Guide To Truck Injury Attorneys In 2023 attorney for truck accident; Wayne,
13 Things About Upvc Tilt And Turn Windows You May Never Have Known tilt and turn Window repairs near me
How Top Performers Use Built-In Technology To Stick To
Top 다바오홀덤
Golf – 7 Strategies To Help Select Your Golf Resort
Destination HiOP
Tips Perform Video Slots Game 에볼루션 블랙잭 조작
Airlie Beach Tours – Different Airlie Beach Tours You Must Experience!
광주알밤
Here’s An Interesting Fact About Truck Accident Attorney.
Truck Accident Attorney big Truck accident lawyer
Great site you’ve got here.. It’s difficult to find high quality writing like yours these days.
I truly appreciate people like you! Take care!!
Windows Doctor Tools To Ease Your Everyday Lifethe Only Windows Doctor Technique Every Person Needs To
Know Windows Doctor
Does Discount Massage Hurt Your Business Model? 광주유흥
What Chiropractor Treatments Are Present? 오피
Great site you have got here.. It’s difficult to find high
quality writing like yours nowadays. I honestly appreciate people
like you! Take care!!
Singles Bar 오피 (https://amlsing.com/)
Who Is Truck Accidents Lawyers Near Me And Why You
Should Consider Truck Accidents Lawyers Near Me Semi truck Accident attorney near me
I all the time emailed this website post page to all my
contacts, for the reason that if like to read it next
my contacts will too.
How To Spot A Quality Massage And Spa Service 광주유흥 (Aracely)
The Best Asbestos Mesothelioma Lawsuit Tips To Transform Your Life Asbestos Lawsuit
Pretty nice post. I simply stumbled upon your weblog and wanted to
mention that I have really loved browsing your
blog posts. After all I’ll be subscribing on your feed and I
hope you write again soon!
Private Club 강남오피 (Luann)
Bangkok – An Ideal Tourist Destination 하이오피
Very nice write-up. I definitely appreciate this website.
Stick with it!
Tips To Steer Your Through New York At Night 광주마사지
(Leonard)
Party Planning – 3 Tips Aid You Plan hiop
If some one needs to be updated with most recent technologies after that he must be visit this site and be up to date
everyday.
Beaches Consider Before Booking Thailand Hotels 광주키스방
The Ultimate Relaxation – A Hot Bath 부달
Making Choices – Online Massage Therapy 오피 (Emerson)
What’s The Job Market For Affordable SEO Agency Professionals?
affordable Seo agency
My brother suggested I would possibly like this website.
He was entirely right. This post truly made my day.
You cann’t consider simply how a lot time I had spent
for this information! Thank you!
Tips On How To Save At Your Marriage 오피
5 Killer Quora Questions On Big American Fridge Freezer
big American Fridge freezers
Searching For Inspiration? Try Looking Up Car Accident car accident Injury Attorneys
How November 23 Big With Mobile Marketing For Up-And-Coming
Small To Large Businesses 다바오4989 (http://www2.saganet.ne.jp/cgi-bin/hatto/board/wwwboard.pl?l=w)
Why Attorneys For Mesothelioma Is Your Next Big Obsession Mesothelioma lawsuit
Bad Credit Personal Loans – Bbb Helps Those Desperate In Need Of Assistance
다바오
Hmm it seems like your blog ate my first comment (it was
super long) so I guess I’ll just sum it up what I
had written and say, I’m thoroughly enjoying your blog. I too
am an aspiring blog writer but I’m still new to everything.
Do you have any tips for first-time blog
writers? I’d genuinely appreciate it.
Using Your Hot Tub For Ultimate Relaxation Budal
Why You Should Focus On Improving Audi A4 Key Replacement Replace audi key Fob
Taking A Calming Visit To Your Spa 하이오피
15 Up-And-Coming 50 50 American Fridge Freezer Bloggers
You Need To Watch freestanding american fridge freezer
(Estela)
Responsible For The Hire Auto Accident Lawyer Budget?
12 Top Ways To Spend Your Money car Wreck Lawyers near me
Get Children Out The Doorway On Quantity Of The Morning With These 9 Tips 서울유흥
That is a great tip especially to those new to the blogosphere.
Simple but very precise info… Many thanks for sharing this
one. A must read article!
Calming Art Of Massage 대전유흥
Rent Chicago Party Bus Services For All Your Bachelor Party 하이오피
How Location On The End-Of-The-Year Work Party 광주키스방 (Angelica)
How To Travel For Free By Leading Or Promoting Tours 유흥
This Is The Ultimate Guide To Trucking Accident Attorneys truck accident attorneys near me – Houston –
Your means of describing the whole thing in this paragraph is actually
good, every one be capable of without difficulty understand it,
Thanks a lot.
Professional SEO Company Tips From The Top In The Business business
Pretty section of content. I just stumbled upon your site and in accession capital to assert
that I acquire in fact enjoyed account your blog
posts. Any way I’ll be subscribing to your feeds and even I achievement you access consistently fast.
Nightlife In Patong As Well As The Rest Of Phuket 광주유흥
Top Frauds Choosing A Health Spa 광주키스방
Amazing! This blog looks exactly like my old one!
It’s on a totally different subject but it has pretty much the same page layout and design. Wonderful choice of colors!
Titan’s First Spa Swimming Session budal
Thank you ever so for you article post.Thanks Again. Awesome.
10 Best Mobile Apps For Asbestos Mesothelioma Lawyers mesothelioma attorneys (hangoutshelp.net)
Nightlife 잠실오피
This blog was… how do you say it? Relevant!!
Finally I have found something which helped me. Thank you!
How To Utilise For Credit Rating Credit Cards goldpay
17 Reasons You Shouldn’t Not Ignore Hiring Auto Accident Attorney Accident attorney car
The Truth About Unique Wedding Reception Venues – Knowledge Is Power 광주유흥
Cocktail Lounge 제주유흥 (39.104.63.123)
10 Some Tips! Getting The Out Of The Wedding Entertainment 하이오피주소
Tips For Selecting A Spa 광주알밤 (http://www.Hirlevel.wawona.hu/getstat/url/?id=158777&maildate=2011-12-0623:00:02&mailid=80&url=http://mebel-avgust.Ru/otzyvy-klientov.html?view=vitabook)
Make Money Online – 10 Steps To Follow For Setting Up Your Very Own Online Business 다바오충전, Agueda,
Nimes Tourist Attractions – Eurostar To Nimes, Shopping,
Nightlife And Accommodation 아이러브밤
Today, while I was at work, my cousin stole my iPad and tested
to see if it can survive a 30 foot drop, just so she can be a
youtube sensation. My apple ipad is now destroyed and she has 83 views.
I know this is totally off topic but I had to share
it with someone!
What’s up, this weekend is pleasant in support of me, because this time i am reading this enormous
informative piece of writing here at my home.
How To Offer A Home Massage 오피
Adult Entertainment 역삼오피
How To Create A Great Income With Blogs & Advertising And Marketing 다바오포커
The Staples Center – A Sports And Entertainment Mecca!
HiOP
Flights To Perth – For A Delightful Vacation 출장마사지
Some Stupid Ways To Max From The Credit Card 다바오충전 (Marita)
Get Convey . Your Knowledge Unsecured Loans Online goldpay
Hey very interesting blog!
Hey! Do you use Twitter? I’d like to follow you if
that would be ok. I’m absolutely enjoying your blog and look forward to new updates.
I love your blog.. very nice colors & theme.
Did you create this website yourself or did you
hire someone to do it for you? Plz reply as I’m looking to construct my own blog and would like to find out where u
got this from. thank you
New York City Tourism Industry Boosts Newark Hotel
Market 오피; https://Www.Spraylock.Spraylockcp.Com/Download.Php?File=Http://Www.Aha.Ru/~Fordmax/Wwwboard/Messages/324.Html?Url=Http://Tinylink.In/Vapevisionhealthbenefits5765,
Exciting Venues With Essential Conference Facilities 부달
Hmm it appears like your website ate my first comment
(it was extremely long) so I guess I’ll just sum it
up what I had written and say, I’m thoroughly enjoying your blog.
I as well am an aspiring blog writer but I’m still new to
everything. Do you have any tips for first-time blog writers?
I’d certainly appreciate it.
It’s going to be ending of mine day, but before ending I am reading this wonderful piece of writing to increase
my know-how.
A Professional Karaoke System For Property OP
Five Top Day Spa Services To Pamper Yourself With 하이오피사이트
Say, you got a nice article post.Much thanks again. Cool.
Surviving The Rat Race By Utilizing Stress Management Techniques
유흥
Very shortly this site will be famous among all blog viewers,
due to it’s pleasant content
What City Should I Move Which Will? 광주마사지
Why Retro Small Fridge Freezer May Be More Dangerous Than You Realized american fridge Freezer retro
Very informative article post.Really looking forward to read more. Cool.
Actually when someone doesn’t be aware of after that its up to other people that
they will assist, so here it takes place.
I think that is among the most important info for me.
And i’m happy reading your article. However wanna commentary on few normal issues, The site style is wonderful,
the articles is in point of fact excellent : D.
Just right activity, cheers
The Luxury Of Luxury Bathtubs – Why These Are Worth
It 부산
Effective Fitness Marketing Techniques 출장
Party Scene 선릉오피
How Establish Your Own Hot Stone Massage Kit 테라피
It’s a pity you don’t have a donate button! I’d certainly donate to this brilliant blog!
I guess for now i’ll settle for book-marking and adding your RSS feed to my Google account.
I look forward to new updates and will share this website with my Facebook group.
Talk soon!
It’s wonderful that you are getting ideas from this paragraph as well as from our argument made at this time.
A big thank you for your blog post.Really looking forward to read more. Keep writing.
What Is Suzuki Swift Remote Key Replacement And Why
Is Everyone Talking About It? suzuki swift smart key programming (timeoftheworld.Date)
Really appreciate you sharing this article post.Really thank you!
Professional Karaoke Machines – Buying The Optimal
One For Your Special Needs OP
The Reasons Order New Driver’s License Is Everywhere This Year beställa körkort internationalt körkort
I value the blog.Thanks Again. Awesome.
Thank you a bunch for sharing this with all of us you actually recognize what you
are talking about! Bookmarked. Please also consult with my web
site =). We will have a hyperlink exchange arrangement among us
Rent Chicago Party Bus Services For Use In Your Bachelor Party 테라피
Lounge Bar 하이오피주소
10 Good Book Workplace Christmas Party Today 오피, http://Www.Coles-Directory.Com,
Hi there Dear, are you in fact visiting this web page regularly, if so after that you will without doubt obtain good know-how.
Sony Ericsson C905 Accessories – Essentials For Your Phone 다바오충전
Planning A Stag And Doe Party On A Spending Budget
광주마사지
I read this article fully about the resemblance of
most recent and earlier technologies, it’s remarkable article.
Co-Ed Baby Showers: Proficient Event That Everyone Can Enjoy HiOP
Peculiar article, totally what I needed.
I enjoy what you guys tend to be up too. This type of clever
work and exposure! Keep up the very good works guys I’ve added you guys to my personal blogroll.
Las Vegas Nightclubs – Vip Hosting And A Wide Range Of Music 인천유흥
5 Killer Quora Answers On Pushchairs Car Seats pushchairs car Seats
The Most Significant Issue With What Is The Average Payout For Asbestos,
And How You Can Repair It Asbestos Lawsuits
Burlesque Show op
5 Killer Quora Answers On Legal Representation For Birth Injuries Good Injury Lawyers Near Me
Hey there, I think your website might be having browser compatibility
issues. When I look at your blog site in Safari,
it looks fine but when opening in Internet Explorer, it has some overlapping.
I just wanted to give you a quick heads up! Other then that, wonderful blog!
Simply desire to say your article is as amazing. The clarity in your post is simply excellent and
i can assume you’re an expert on this subject.
Well with your permission allow me to grab your RSS feed to keep up to date with forthcoming post.
Thanks a million and please continue the rewarding work.
Howdy! Do you know if they make any plugins to assist
with SEO? I’m trying to get my blog to rank for some targeted
keywords but I’m not seeing very good success. If you
know of any please share. Kudos!
Discovering Nightlife In Marrakech 밤문화 – Prince,
Mother’s Day Gift Ideas – For Teenagers 광주오피
In your opinion, what are the main pros and cons of this product?Regard IT Telkom
Private Party 역삼오피 (Jennie)
What i do not realize is in fact how you are now not really a lot
more neatly-favored than you may be now. You’re so
intelligent. You know thus significantly on the subject of this matter, produced
me individually imagine it from so many varied angles.
Its like men and women don’t seem to be interested unless it is one thing to
do with Lady gaga! Your personal stuffs outstanding.
All the time handle it up!
The 10 Most Terrifying Things About Retro Fridge Freezers
Uk retro fridge freezers uk, Brandie,
10 Places That You Can Find Asbestos Exposure Compensation asbestos lawyers
You Can Explain Asbestos Poisoning Compensation To Your Mom Asbestos lawsuit
What’s The Current Job Market For Tilt And Turn Window Handles Uk Professionals
Like? Tilt And Turn Window Handles
I’m really impressed along with your writing talents and also with the format
to your blog. Is this a paid subject matter or did
you modify it your self? Either way keep up the excellent quality writing, it is rare to look
a nice blog like this one these days..
Your First Trip Towards Spa – What To Try To To And What To Anticipate 부산달리기
An Great Way To Get Your Massage Business Found On The Internet 광주키스방
Don’t Compromise When It Comes To Buying Massage Therapy Equipment 알밤; Beulah,
What’s The Current Job Market For Accident Lawyers Baton Rouge Professionals?
accident Lawyer (gaines-johannsen-3.technetbloggers.de)
Singles Bar 역삼오피
Back Injury Attorneys Near Me Tools To Make Your Daily Life Back Injury Attorneys Near Me Trick Every Individual Should Be Able To
injury attorneys Near me
20 Things That Only The Most Devoted Mesothelioma Asbestos Claim Fans Know Mesothelioma Lawyer
Hello! Do you know if they make any plugins to safeguard against hackers?
I’m kinda paranoid about losing everything I’ve worked hard on. Any recommendations?
Five Secrets To Arrange An Eye-Catching Family Reunion Party 서울유흥 (39.104.63.123)
Speakeasy OP
A Step-By’-Step Guide To Picking Your Asbestos
Mesothelioma Lawsuit Asbestos Lawyers
How Acquire The Ultimate Birthday Party Places 광주키스방 – melody.lib.net –
Thanks for another informative site. The place
else could I get that kind of information written in such a perfect
method? I’ve a challenge that I’m simply now working on, and I have been at the look out
for such info.
Massage Therapist Salary HiOP
Awesome! Its in fact amazing post, I have got much clear idea on the topic
of from this post.
Have you ever thought about writing an ebook or guest authoring on other sites?
I have a blog centered on the same information you discuss and would really
like to have you share some stories/information. I know my viewers would enjoy your
work. If you’re even remotely interested, feel free to shoot me an e-mail.
20 Myths About Ny Asbestos Litigation: Dispelled Asbestos Lawyers
Magnificent beat ! I wish to apprentice while you amend your site, how can i subscribe for a blog web
site? The account aided me a acceptable deal. I had been tiny bit acquainted
of this your broadcast provided bright clear idea
My coder is trying to convince me to move to .net from
PHP. I have always disliked the idea because of the costs.
But he’s tryiong none the less. I’ve been using Movable-type on a variety of websites
for about a year and am concerned about switching to another
platform. I have heard great things about blogengine.net.
Is there a way I can transfer all my wordpress posts into it?
Any kind of help would be really appreciated!
Full Service Spa OP
Why Buy A Driving License Is The Best Choice For You?
Kupie Prawo jazdy
Hi there! Someone in my Facebook group shared this site with us so I came to
take a look. I’m definitely enjoying the information. I’m book-marking and will be
tweeting this to my followers! Outstanding blog and terrific design and style.
It’s awesome in favor of me to have a web site, which is valuable designed for my knowledge.
thanks admin
What’s The Point Of Nobody Caring About Houston Asbestos Attorney Mesothelioma Lawyers (Hefeiyechang.Com)
The Ultimate Glossary For Terms Related To Asbestos Cancer Lawyer Mesothelioma Settlement Mesothelioma Lawyers
What Is Misted Up Double Glazing? History Of Misted Up Double
Glazing In 10 Milestones Double Glazing Misting
10 Inspirational Graphics About Accident Attorney attorney accident lawyer (trade-britanica.Trade)
A How-To Guide For Asbestos Cancer Lawsuit Lawyer Mesothelioma
From Beginning To End mesothelioma attorney (Mai)
I do not even know how I ended up here, but I thought this post was great.
I don’t know who you are but certainly you are going to a famous blogger if you aren’t already 😉 Cheers!
12 Companies Leading The Way In Good Accident Lawyers
good accident Lawyers near me
You actually make it seem so easy with your presentation but
I find this matter to be really something which I think I would
never understand. It seems too complicated and extremely broad for me.
I’m looking forward for your next post, I’ll try to get the hang of it!
The Unknown Benefits Of Locksmith Services locksmith services Near me
You can certainly see your skills within the article you write.
The world hopes for even more passionate writers such
as you who aren’t afraid to say how they believe. Always go after your heart.
I’m truly enjoying the design and layout of your blog.
It’s a very easy on the eyes which makes it much more
enjoyable for me to come here and visit more often. Did you hire out a developer to create your
theme? Great work!
Hi there, yeah this piece of writing is genuinely good and I have learned lot of things from
it on the topic of blogging. thanks.
Why You Should Focus On Enhancing French Door Windows french doors and
windows – jinrihuodong.Com –
What NOT To Do In The Door Doctor Near Me Industry The door doctor near me
5 Reasons To Be An Online Buy A Driving License Shop And 5 Reasons Not To
wie kann ich meinen führerschein kaufen (Dennis)
Hi there very nice website!! Man .. Excellent ..
Wonderful .. I’ll bookmark your site and take the feeds also?
I’m happy to seek out a lot of useful information here in the publish, we’d like develop more strategies in this regard,
thank you for sharing. . . . . .
Tae Kwon Do – The Involving Kicking And Punching 웹툰사이트 (https://click4r.com/)
A Step-By-Step Guide To Adults Toys Shop From Beginning To End adults Sex Toys
9 Things Your Parents Teach You About Buy UK Driving License Without Test buy uk Driving license without test
I feel this is among the most important info for
me. And i’m glad studying your article. However want to observation on few
basic things, The web site style is wonderful, the articles
is really nice : D. Excellent job, cheers
Brand Yourself Publishing Online – Top Tips 웹툰미리보기
Cause Of Hair Decrease In Women – The Role Of Dht & Sebum 웹툰
Travel Advice For South East Asia 주소모음
10 Undeniable Reasons People Hate Upvc Windows & Doors upvc
doors with windows (Parthenia)
How Popular Are You,. Online The Actual? 웹툰미리보기
Natural Therapy Of Panic Attacks And Anxiety 마사지
The Worst Advice We’ve Ever Been Given About Buy Am Driving License Online KöPa Ett KöRkort
How To Travel Between Cities In Vietnam 하노이밤문화
5 Top Reasons To Visit South Korea 웹툰
See What Gas Service Engineer Near Me Tricks The Celebs Are
Using Gas service engineer near me
I was recommended this blog by my cousin. I’m not sure whether this post
is written by him as no one else know such detailed about my difficulty.
You’re amazing! Thanks!
8 Quick Ways To Unwind Before It Really Is Date 유흥사이트
It’s not my first time to pay a quick visit this
web page, i am browsing this web page dailly and obtain nice data from here
daily.
Locations For A Beach Wedding 출장마사지
Proven Methodologies For Conquering Stress 유흥사이트
Where Can You Get The Top Adult Toys Information? Bunny Adult toy
Link exchange is nothing else but it is
only placing the other person’s website link on your page at suitable place and
other person will also do similar in favor of you.
Can you tell us more about this? I’d love to find out more
details.
20 Fun Facts About Apply For A New Drivers License beställa Nytt skoterkort
How To Travel In Safety And Style With Personal Occasions Limo Rental Service 오피사이트 (Dawna)
All About New York City Bars HiOP
Networking For Career Success – Create An Quality Inventory
하이오피
The 10 Most Worst Drivers License Order New-Related FAILS Of All
Time Could Have Been Prevented nytt körkort borttappat
What’s The Current Job Market For Why Are The Glaceous Macaw And Hyancith Macaw So Alike Professionals?
why are the glaceous macaw and hyancith macaw so alike (Myles)
What Experts In The Field Would Like You To Learn kupie prawo jazdy kat
b (bbs.Lingshangkaihua.com)
Adults Toys Shop Tips To Relax Your Daily Life Adults Toys Shop Trick That
Everyone Should Learn adults toys shop (Sherman)
This is my first time pay a visit at here and
i am really impressed to read everthing at single place.
Burlesque Show 하이오피사이트
9 Lessons Your Parents Taught You About Buy A Full UK Driving Licence Buy A Full Uk Driving Licence
5 Buy UK Driving License Projects For Every Budget fake uk driving license
Hey! Quick question that’s entirely off topic.
Do you know how to make your site mobile friendly? My site
looks weird when viewing from my iphone4. I’m trying to find a theme or plugin that might be able to correct this
issue. If you have any suggestions, please share. Many thanks!
Five Great European Holiday Destinations 하이오피주소
Travel Advice For East Asia 베트남유흥
Top Golf Equipment In The United Kingdom OP
Event Planning, Planning Opertation That Is Actually Going
To Remembered OP
Can You Are Converting Your Marketing Into A Religion? 오피
15 Interesting Facts About New Drivers License That You’d Never Been Educated About BestäLla Nytt KöRkort – http://Www.Xuetu123.Com/Home.Php?Mod=Space&Uid=10282288,
You really make it seem so easy with your presentation but
I find this matter to be really something which I think I would never
understand. It seems too complicated and extremely broad
for me. I am looking forward for your next post, I’ll try to
get the hang of it!
Top 3 Tips For Planning Preferred Myrtle Beach Vacation OP
5 Laws Anybody Working In Private Mental Health Assessment Should Be Aware Of mental Health assessment for Schizophrenia
20 Tools That Will Make You More Successful At Sex Doll Ultra Realistic How much
is a Realistic sex doll [bookmarkzones.trade]
Buy Driving License A1 101 This Is The Ultimate Guide
For Beginners kup prawo Jazdy polska (https://edu.dobro.ru/bitrix/redirect.php?Goto=https://prawo-jazdy-Wyszkow.pl)
Guide To African Grey Parrots Sale: The Intermediate Guide Towards African Grey Parrots Sale african Grey parrot
10 Best Mobile Apps For Germany For Buying A Driving License füHrerschein kaufen ohne prüfung
Preshow Planning Equals Success : 10 Essential
Questions You Require To Ask 하이오피사이트
20 Reasons To Believe Gas Safety Certificate Near Me Will Never Be Forgotten gas safety certificates (hubgit.cn)
3 Ways In Which The SEO Tools Software Influences Your Life
What Are Seo Tools
Tips Come Across A Masseuse Job 부산달리기
This website really has all of the information I needed about this subject
and didn’t know who to ask.
9 . What Your Parents Taught You About Content Marketing For B2b content Marketing for b2b (https://Kingranks.com)
The 9 Things Your Parents Teach You About New Wood Pallet For Sale new
wood pallet for sale (https://muse.union.edu/)
Forget Buy A Driving License In Germany: 10 Reasons Why You Do Not Need It Legalen FüHrerschein Kaufen
What’s The Reason Adults Toys Shop Is Fast Becoming The Hottest Trend For 2024 masturbation Adult toy
deal – http://www.tianxiaputao.Com –
What’s The Job Market For Buy UK Driving License Professionals?
buy uk driving license
Are Buy A Driving License Legally The Best Thing
There Ever Was? Kann man führerschein kaufen; http://www.kaseisyoji.com,
Ask Me Anything: 10 Responses To Your Questions About Double
Buggy Tandem Double Stroller
Psychiatry Assessment Tools To Improve Your Daily Lifethe One Psychiatry
Assessment Trick Every Individual Should Learn Psychiatry Assessment
How To Save Money On Scooter Driving License prawo jazdy c+e
What’s The Current Job Market For Talking African Grey Parrot For Sale Professionals Like?
african grey parrot for sale (https://hackett-wilhelmsen.mdwrite.net)
Hurrah, that’s what I was looking for, what a information! present here at this
website, thanks admin of this web site.
How Egg Adult Toy Was The Most Talked About Trend Of 2024 app
controlled adult toys (Georgianna)
7 Small Changes That Will Make A Big Difference With Your Buy UK Drivers License buy uk drivers
License online (ceshi.xyhero.com)
8 Tips To Enhance Your Private Psychiatrist Durham Game Private psychiatrist uk Cost
7 Essential Tips For Making The Most Out Of Your Buy
A German Driving License Legally Registrierten Führerschein kaufen
10 Easy Ways To Figure Out Your Buy Wheel Loader Driving License Online de körkort
It’s Enough! 15 Things About How Much Does A Scooter Driving License Cost We’re
Sick Of Hearing prawo Jazdy A1
What Is The Window Doctors Term And How To Use It the Window doctors
What’s Happening i am new to this, I stumbled upon this I have
discovered It absolutely useful and it has aided me out loads.
I hope to contribute & assist different users like
its helped me. Great job.
5 Upvc Door Hinges Replacement Projects For Every Budget upvc windows hinges
3 Common Reasons Why Your Buy A Driving License Without Advance Payment Isn’t Working (And How To Fix It)
eu führerschein kaufen erfahrungen (Dora)
It’s Time To Expand Your Pram Pushchair 2 In 1 Options tandem Stroller
20 Resources To Help You Become Better At Buy Tilt And Turn Windows tilt and turn Windows online – tenniscave32.werite.net,
This Is The Ultimate Guide To Upvc Door Hinge Repair Near Me
Friction Hinges For Aluminium Windows (Playplot2.Werite.Net)
Double Pushchair Sale: Myths And Facts Behind Double Pushchair Sale Double Stroller Sale
The People Who Are Closest To Heating Engineer In Buckingham Share Some Big
Secrets Gas Safety Engineer Buckingham
Who’s The Top Expert In The World On Three Wheeled Buggies?
3 wheel buggy (Darnell)
What’s The Job Market For Window Repairs Luton Professionals Like?
window repairs luton; George,
The 10 Scariest Things About Heating And Gas Engineer heating
and gas engineer (Taj)
10 Inspirational Graphics About Upvc Patio Door
Repairs Repair Crack In Upvc Door (https://Anotepad.Com/Notes/X7Hcmpd7)
Very rapidly this web page will be famous among all blogging viewers,
due to it’s nice posts
10 Facebook Pages That Are The Best Of All Time Concerning Boiler Repairs
Milton Keynes gas safety certificate milton keynes Online
Five Things You’ve Never Learned About Gas Safety Checks In Buckingham Landlord gas safety certificate Buckingham
Why Double Glazed Units Near Me Is A Must At Least Once In Your Lifetime replacement double glazed units
near me (Rosalie)
11 Ways To Destroy Your Window Hinge Repairs Near Me Double Glazed Window Hinge
The 10 Scariest Things About Window Repairman Near Me window repairman near me
The 10 Scariest Things About Double Glazing Seal Repairs replacement window
seals near me [Maggie]
This Is How Tilt Turn Window Handles Will
Look Like In 10 Years Time old aluminium window handles; Kenneth,
Replacing Window Seal: 11 Things You’re Leaving Out upvc sealed unit replacements (Beau)
10 Reasons That People Are Hateful To Psychiatrist Assessment Near Me Psychiatrist Assessment Near Me Psychiatry uk assessment
What’s The Current Job Market For Flush Sash Windows Professionals?
Flush Sash windows
The Most Common Double Glazed Window Handles Mistake
Every Beginner Makes replacement window handles
near me, http://idea.informer.com/,
The 9 Things Your Parents Taught You About Misted Window Repair Cost Misted Window repair Cost
I am really loving the theme/design of your
website. Do you ever run into any internet browser compatibility issues?
A number of my blog visitors have complained about my site not working correctly in Explorer
but looks great in Opera. Do you have any ideas to help
fix this problem?
From Around The Web 20 Amazing Infographics About Sash Window Restoration sash style double glazed windows
10 Things That Your Family Taught You About Psychiatrist Assessment UK psychiatrist Assessment uk
The 10 Most Terrifying Things About Compact Double Buggy
compact double buggy, Vito,
How The 10 Most Disastrous Upvc Door Panel Replacement Fails
Of All Time Could Have Been Prevented upvc door panel replacement cat
flap – Junior,
How To Explain General Psychiatric Assessment To Your Grandparents psychiatric patient assessment (http://www.bioguiden.se)
Pay Attention: Watch Out For How Replacement
Window Hinges Is Taking Over And What Can We Do About It Pvc window hinges
15 Gifts For The Replace Lock In Upvc Door Lover In Your Life replacement upvc window locks
Hello everyone, it’s my first go to see at this web site, and paragraph
is genuinely fruitful for me, keep up posting these types of
posts.
7 Small Changes That Will Make The Difference With Your Replace Lock Upvc Door window
and door lock repair (https://pattern-wiki.win/wiki/How_Upvc_Door_Locks_Replacement_Became_The_Hottest_Trend_Of_2023)
15 Terms Everybody Within The Double Glazing Window Repairs Industry
Should Know repairs to upvc doors
You’ll Be Unable To Guess Upvc Window Repairs Near Me’s Benefits
upvc window repairs near me
11 Creative Ways To Write About UPVC Door Lock replacing window locks
The Top Reasons People Succeed On The Private Psychiatrist Assessment Near Me Industry private psychiatrist near me uk
I am actually delighted to read this webpage posts which carries plenty of
valuable data, thanks for providing these kinds of information.
See What Window Mechanism Repair Near Me Tricks The Celebs Are Making Use Of Window Mechanism Repair Near Me
A The Complete Guide To Double Glazing Offers Near Me From Start To Finish double galzing
What Is Milton Keynes Door And Window And How To Use
What Is Milton Keynes Door And Window And How To Use doors And Windows milton keynes,
suhr-contreras.mdwrite.net,
What you said was actually very reasonable. However, what about this?
what if you were to write a awesome post title? I am not saying your information isn’t solid,
however suppose you added a title to maybe get a person’s attention? I mean 医薬におけるバリデーション | お役立ち情報 is kinda boring.
You could peek at Yahoo’s home page and note how they create post titles to grab
people to click. You might add a video or a picture or two to get readers excited about what you’ve
written. Just my opinion, it might make your blog a little livelier.
The Best Place To Research Psychiatric Assessment For Court Online urgent psychiatric assessment
See What Double Glazed Windows Repairs Tricks The Celebs Are
Using double Glazed Windows repairs
7 Effective Tips To Make The Most Of Your Lock Replacement
Upvc Door Upvc Windows Locks Repair
You’ll Never Guess This Double Glazed Window Handles’s Secrets Double Glazed window handles
Don’t Stop! 15 Things About Double Glazed Windows Bromley We’re Sick
Of Hearing Front Doors Bromley
Thanks , I’ve recently been looking for info approximately this subject for a while
and yours is the greatest I’ve found out so far. But, what
in regards to the conclusion? Are you positive in regards
to the supply?
14 Cartoons About Mental Health Tests That Will Brighten Your Day mental
health assessment online (https://telegra.ph/5-Laws-That-Can-Benefit-The-Mental-Health-Assessment-Test-Industry-06-23)
Solutions To Problems With Psychiatrist In Near Me best adhd psychiatrist near me (Chang)
The No. 1 Question That Anyone Working In Mental Health Assessments Needs To Know
How To Answer private mental health diagnosis uk – Sherri,
Assessment Mental Health Techniques To Simplify Your Everyday Lifethe
Only Assessment Mental Health Trick That Should Be Used By Everyone Know assessment
mental health (Rosalyn)
You’ll Never Guess This Repair Window Handle’s Secrets repair window
handle (marvelvsdc.faith)
Are You Sick Of Mental Health Assessments? 10 Sources Of Inspiration That’ll Invigorate Your Love Assessment For mental health
The 10 Most Scariest Things About Mental Health Assessments mental
health screening online (Warner)
That is very fascinating, You are an overly skilled blogger.
I have joined your rss feed and stay up for in quest of extra of your great post.
Additionally, I have shared your website in my social networks
It’s really very complex in this busy life to listen news on TV, therefore
I simply use world wide web for that purpose, and take
the latest news.
Double Glazing Near Me Tips To Relax Your Daily Lifethe One Double Glazing
Near Me Trick That Everyone Should Be Able To Double glazing near Me
Sightcare has actually been a fantastic addition to my wellness regimen. I began taking it to attend to the eye stress I experienced from long hours of screen time, and it has actually worked marvels. My vision is sharper, and I no much longer really feel the pain I used to. The all-natural components provide me tranquility of mind, knowing I’m supporting my eye wellness with no damaging chemicals. Sightcare is a must-have for any individual seeking to maintain healthy vision and lower eye tiredness.
My blog … https://Zapinacz.pl/@anastasia29777?page=about
Yes! Finally someone writes about gobigguru.com.
I’ve been making use of DentaVim for a couple of months now, and the outcomes are fantastic. My teeth are whiter, my periodontals are healthier, and I no more stress over bad breath. The supplement is easy to take and does not have any type of undesirable aftertaste. I highly suggest DentaVim to any person seeking to enhance their oral health.
Here is my web blog: http://chansolburn.com/bbs/board.php?bo_table=free&wr_id=552005
I’ve been utilizing TonicGreens for a couple of months now, and the results are outstanding! My power levels have actually increased, and I really feel extra focused throughout the day. And also, my immune system really feels more powerful than ever. Extremely suggest this supplement to anyone seeking to enhance their general wellness!
My web-site; http://grainfather.co.nz/employer/tonicgreens-ltd
I’ve attempted several weight-loss supplements, however Nagano Lean Body Tonic stands out. The mix of environment-friendly tea remove, ginger, and turmeric extract is powerful. It has actually helped me manage my hunger and enhance my metabolic rate. Plus, it’s easy to incorporate right into my day-to-day regimen. I’m delighted with the results!
My site – https://www.Laciotatentreprendre.fr/employer/us-naganotonics/
Private Online Psychiatrist: 11 Thing You’re Forgetting To Do private psychiatrist near me uk
10 Places To Find Upvc Door Panels Upvc Door Panel Design (Telegra.Ph)
20 Questions You Must Always To Ask About Replacement Upvc Door Lock Prior To Purchasing Replacement Upvc Door Lock Double glazing window locks replace
2 In 1 Pram System Techniques To Simplify Your Daily Life 2 In 1 Pram System Trick
Every Individual Should Learn 2 In 1 Pram system
Because beginning Nagano Lean Body Tonic, I’ve seen substantial renovations in my digestion and energy levels. The green tea remove and turmeric job wonders. It’s an all-natural and efficient way to support weight monitoring. I’m really happy with the outcomes and will proceed utilizing it.
Feel free to visit my blog http://bolsatrabajo.cusur.Udg.mx/employer/us-naganotonics/
Patong Beach In Thailand 오피사이트
I’ve been making use of DentaVim for a few months now, and the outcomes are amazing. My teeth are whiter, my gum tissues are healthier, and I no much longer worry about halitosis. The supplement is very easy to take and doesn’t have any unpleasant aftertaste. I highly advise DentaVim to anyone looking to enhance their dental wellness.
My web blog: https://classified.Completemarts.com/profile/fletadelagarza
Where Can You Find The Most Reliable Tilt And Turn Windows Mechanism Information? tilt and Turn windows how to open
DentaVim has actually been a game-changer for my oral health and wellness. After just a couple of weeks of use, I noticed a substantial reduction in periodontal swelling and my breath is fresher than ever. The all-natural ingredients give me assurance, recognizing I’m not putting unsafe chemicals right into my body. Extremely suggest!
Take a look at my web page; http://app.vellorepropertybazaar.in/profile/joshuao3448862
5 Laws That’ll Help The Window Handles And Locks Industry types Of window handles
The 10 Scariest Things About Upvc Window Repairs Near Me Window Repairs Near Me
Since I began making use of Provadent, my oral examinations have actually been far better. My teeth are more powerful, and my periodontals are healthier. The all-natural formula is gentle yet reliable. Provadent is now a staple in my daily regimen.
Feel free to surf to my web page https://Soudfa.It5h.com/@kathybook71726
5 Must-Know Replacing Upvc Door Lock Practices For 2023 replacement Window lock
The Reasons Double Glazed Windows Repairs Near Me Isn’t As Easy
As You Think window repair service near me
How To Make An Amazing Instagram Video About Window Handles upvc window handles
near me (https://telegra.ph/Five-Things-You-Dont-Know-About-Window-Handles-Repair-09-18)
Deciding Upon The Location Of One’s Hot Tub 오피
Burdeos and Greunke claim creatine monohydrate can assist construct muscle.
Also visit my webpage :: https://Rapid.tube/@darlenevandive?page=about
DentaVim is an amazing item! My dentist recommended it to help with my periodontal problems, and I’ve seen extraordinary outcomes. My gums are no more inflamed, and my breath stays fresh all the time. It’s simple to take and fits perfectly into my day-to-day regimen. I couldn’t be better with the outcomes.
My web page – https://Theelitejob.com/profile/gitamacaluso36
Reasons To Rent A Wedding Coordinator 동탄오피
Turtle Bay Resort Golf Wedge – Kahuku – Oahu – Hi 창원오피사이트
Adult Mental Health Services: The History Of Adult Mental Health Services In 10 Milestones book a mental health assessment
How To Get Better Results Out Of Your Double Strollers off road double buggy [lovewiki.Faith]
Spice Increase Date Using A Couple’s Massage 유흥
Indisputable Proof You Need Mental Health Test Online mental health screening uk
Avoid Making This Fatal Mistake You’re Using Your Assessment Of A
Psychiatric Patient Psychiatric Assessment edinburgh
Keto Flow has actually been a game-changer for my weight reduction trip. The gummies are not only scrumptious however likewise efficient in helping me melt fat and suppress my appetite. I have actually seen a substantial boost in my energy levels considering that I started taking them. If you’re trying to find a reputable keto supplement, Keto Flow is the way to go!
Also visit my web blog: https://classified.completemarts.com/profile/celsacramp067
20 Fun Details About Psychiatric Assessment UK psychiatry-uk Adhd self assessment
15 Best 3 Wheel Travel System Bloggers You Must Follow best 3 wheel travel
system (Lucie)
VisiSoothe has actually made an obvious difference in my vision. I made use of to deal with blurred vision and eye stress, yet given that taking VisiSoothe, my eyes feel extra unwinded and concentrated. The all-natural active ingredients offer me comfort, understanding I’m sustaining my eye health safely. VisiSoothe is an outstanding choice for any person looking to enhance their vision naturally.
Feel free to surf to my web site: https://Holisticrecruiters.uk/employer/visisoothe-web/
Stress Relief Massage – Stress Management
Through Therapeutic Massage 부천오피사이트
It’s really a cool and helpful piece of information. I’m satisfied that you just shared this useful information with us.
Please keep us informed like this. Thank you for sharing.
Assessment Mental Health Tools To Streamline Your Everyday Lifethe
Only Assessment Mental Health Trick That Should Be Used By Everyone Be Able To assessment mental health, Rosemarie,
Creating Muscles Weight Gain 오피 (pattern-wiki.win)
Good way of describing, and good piece of writing to take data about my presentation focus, which
i am going to convey in institution of higher education.
How To Explain Mental Health UK To Your Grandparents Mental Health Test Uk
Why Live Wedding Party Bands End Up Being Best 구미오피사이트
I was hesitant at initially, but ProDentim surpassed my expectations. My teeth are whiter, and my gum tissues are healthier. The all-natural, non-GMO components offer me comfort. ProDentim is now a staple in my daily oral treatment routine.
My web page :: http://Elevatepalestine.com/employer/sup-prodentim
What’s The Current Job Market For Glass Seal Repair Professionals?
glass seal repair, Bianca,
What Window Hinges Experts Want You To Know? fix window hinge (Charmain)
9 Lessons Your Parents Taught You About Upvc Double Glazed Sash Windows double Glazed sash
Window – botdb.Win –
11 Strategies To Completely Defy Your Mental Health Assessment
mental Health diagnostic assessment
Wisdom On Basic Psychiatric Assessment From An Older Five-Year-Old psychiatric assessment online uk (Sandy)
How To Create An Awesome Instagram Video About Psychiatric Assessment UK
how to get A psychiatric assessment; http://www.maanation.com,
The Reasons To Work On This Upvc Door Locking Mechanism double glazing window locks
repairs (menwiki.men)
I have actually been utilizing ElectroSlim for a month currently, and the results are extraordinary! This electrolyte fat-burning beverage has actually significantly boosted my metabolic rate and aided me lost those stubborn pounds. I love that it’s made with all-natural, non-GMO, and gluten-free components, making it a healthy selection for weight loss. Plus, it keeps me moisturized and invigorated throughout the day. Very advise ElectroSlim to any person aiming to shed weight efficiently!
My webpage – https://Git.Tx.pl/poppyparr7622/katlyn2008/wiki/The+Function+of+Tidy+Consuming+in+Fat+Burning+and+General+Health
How To Plan The Perfect Videoke Party At Home 울산유흥
8 Pitfalls Of Managing Your Own Family Based Business
And How To Overcome Them 하이오피사이트
The 10 Scariest Things About Mental Health Assesment mental Health assesment
Stop Panic Attacks Now And Forever 원주오피
Wow, that’s what I was looking for, what a stuff! existing here at this weblog,
thanks admin of this site.
Are You Getting The Most You Private Psychiatrists?
How Much Is A Private Psychiatrist Uk
Insomnia – What Moment Has Come And Tips On How To Manage It 오피
Hi there! Do you use Twitter? I’d like to follow you if that would be okay.
I’m undoubtedly enjoying your blog and look forward to new posts.
I’m Deeply In Love With Network Marketing! 오피 (renitdaniel.com)
Anger Management – Ways Of Dealing With Unhealthy Anger 인천오피 (https://sciencewiki.science/wiki/American_Registry_For_Internet_Numbers)
15 Interesting Facts About Upvc Door Locks Repair You’ve Never Heard Of repair hole in upvc door
Vip Janitorial Services – Let Them Clean Your Current Act 하이오피사이트
DentaVim has actually been a terrific enhancement to my oral treatment regimen. My gums are no much longer aching, and my breath stays fresh throughout the day. The natural components are gentle yet efficient, and I enjoy that it’s made in an FDA-registered facility. This product genuinely provides on its guarantees.
Feel free to visit my site; https://Dream-Weaver.CO.Kr/bbs/board.php?bo_table=free&wr_id=1511559
I must thank you for the efforts you’ve put in writing this website.
I really hope to check out the same high-grade blog posts by
you in the future as well. In fact, your creative writing abilities has motivated
me to get my own blog now 😉
Hello to every one, it’s truly a nice for me to go to see this site, it consists of important Information.
This paragraph is in fact a pleasant one it assists new web people, who are
wishing in favor of blogging.
Thank you for every other magnificent post.
The place else could anybody get that kind of info in such an ideal manner
of writing? I’ve a presentation next week, and I am at the look for such info.
This is my first time pay a quick visit at here and i am really
happy to read everthing at alone place.
This article provides clear idea for the new visitors of blogging,
that genuinely how to do blogging and site-building.
Nagano Lean Body Tonic has actually been a game-changer for me. I’ve battled with weight monitoring for several years, however this restorative has actually made a noticeable difference. The natural components like green tea remove and turmeric have enhanced my metabolic process and energy levels. I really feel healthier and more certain. Highly advise!
my site – https://escatter11.fullerton.edu/nfs/show_user.php?userid=8022009
I’ve been using VisiSoothe for some time now, and the renovation in my vision is amazing. My eyes really feel much less stretched, and I can see more clearly, even after long hours of screen time. The all-natural formula is a huge plus, and I enjoy that it’s designed to sustain total eye health. VisiSoothe is a wonderful item for anybody wanting to improve their vision and eye health.
Review my page :: https://www.webwiki.fr/doodleordie.com/profile/humordesert8
PrօDeentim has been a lifesaver for my oral health ɑnd wellness.
My teеth are more рowerful, and my gum tissues are no much longer dеlicate.
The natural,non-GMO activе ingredients make it a risk-free choіce for any indіvidual looking to improve their dental health.
I highly advise ProDentim!
my wеb blog Everything You need to know about ProDentim side effects
I’ve discovered a considerable decrease in plaque accumulation considering that I began making use of Provadent. My gum tissues are healthier, and my teeth feel more powerful. The supplement is simple to take, and the outcomes go over. Provadent is a fantastic addition to my dental treatment routine.
Also visit my homepage https://Notes.io/wZirz
Hi there, I enjoy reading all of your article.
I like to write a little comment to support you.
whoah this weblog is wonderful i love reading your
articles. Keep up the good work! You know, many
persons are looking around for this information, you could help them greatly.
Hello! I know this is kinda off topic but I’d figured
I’d ask. Would you be interested in trading links or maybe guest authoring a blog
post or vice-versa? My website discusses a lot
of the same topics as yours and I feel we could greatly benefit
from each other. If you’re interested feel free to shoot me an e-mail.
I look forward to hearing from you! Superb
blog by the way!
There is certainly a lot to find out about this subject. I really like all the points you’ve made.
Also visit my page :: http://hola666.com/home.php?mod=space&uid=1327520
It’s hard to find educated people in this particular topic, but you seem like you know what you’re talking about!
Thanks
How Buy A Driving License Changed Over Time Evolution Of Buy A
Driving License eu führerschein kaufen erfahrungen (Jerold)
This article will help the internet viewers for setting up new web site or even a blog from start to
end.
Your Worst Nightmare Concerning Best Automatic Vacuum Cleaner Be Realized robot vacuum that vacuums and mops
I think what you typed made a lot of sense. But, think about this, what if
you typed a catchier post title? I mean, I don’t want to tell you how
to run your blog, but what if you added a post title to possibly grab a person’s attention? I mean 医薬におけるバリデーション | お役立ち情報 is
a little vanilla. You might look at Yahoo’s front page and
note how they create news titles to get people to open the links.
You might try adding a video or a related picture or two to grab readers excited about what you’ve written. Just my opinion, it would make your website
a little livelier.
The 10 Scariest Things About Best Auto Locksmith
Luton Best Auto Locksmith Luton
20 Things That Only The Most Devoted Luton Best Car Locksmith Fans Know best luton car locksmiths (Robby)
You’ll Never Guess This Best Car Locksmiths Luton’s Secrets
Car locksmiths luton – bbs.0817ch.com,
Guide To Best Car Locksmiths In Luton: The Intermediate Guide On Best
Car Locksmiths In Luton best Car Locksmiths in luton
What Is The Reason Car Locksmith Luton Is The Right Choice For You?
car locksmith in luton (https://marvelvsdc.faith/wiki/10_Best_Auto_Locksmiths_Luton_Meetups_You_Should_Attend)
Why Robot Vacuum Cleaners Uk Is The Right Choice For You?
autonomous vacuum (https://www.pmxwiki.xyz/index.php/User:RyderIvb131493)
How Best Car Locksmiths Luton Has Become The Most Sought-After Trend Of 2024 best car locksmiths
in luton (Luz)
See What Car Locksmith Luton Tricks The Celebs Are Using Car Locksmith Luton
What’s The Job Market For Car Locksmith Luton Professionals?
Car locksmith Luton
A Vibrant Rant About Auto Locksmith Near Luton Best Luton Auto Locksmiths
Five Luton Car Locksmith Projects To Use For Any Budget Best Luton Car locksmiths
A Rewind What People Said About Car Locksmiths
Luton 20 Years Ago luton car Locksmith
5 Things Everyone Gets Wrong Concerning Luton Best Auto Locksmith best Luton auto locksmith
Five People You Must Know In The Car Locksmiths Luton Industry
Best Luton Car Locksmiths
You’ll Be Unable To Guess Best Car Locksmiths Luton’s Tricks car locksmiths luton
See What Luton Car Locksmiths Tricks The Celebs Are Using luton car Locksmiths
You’ll Be Unable To Guess Auto Locksmiths Near Luton’s Benefits Auto Locksmiths Near Luton
How Auto Locksmiths Luton Arose To Be The Top Trend On Social Media best luton Auto Locksmith (imoodle.win)
The Most Common Luton Best Car Locksmith Mistake Every Newbie Makes
best auto locksmiths in luton – xxh5gamebbs.Uwan.Com –
A Look Into Car Locksmiths Near Luton’s Secrets Of Car Locksmiths Near
Luton auto locksmith in luton, Epifania,
9 Things Your Parents Teach You About Automatic Vacuum Cleaners automatic vacuum cleaner,
jsbs.kr,
The 10 Scariest Things About Windows And Doors Replacement windows and doors replacement
(Velva)
I am really impressed with your writing skills as well as with the layout on your
weblog. Is this a paid theme or did you modify it yourself?
Anyway keep up the excellent quality writing, it is rare to see a nice blog like this one today.
See What Front Door And Window Tricks The Celebs Are Making Use Of front door and window (Karen)
See What Window In Door Tricks The Celebs Are Using window In Door
See What Window Upvc Door Tricks The Celebs Are Using window upvc door
You’ll Never Guess This New Windows And Doors’s Tricks new windows and Doors
5 Killer Quora Answers On French Windows And Doors french
Windows And doors (http://Www.Seoulschool.org)
You’ll Be Unable To Guess Bedside Cot Bed’s Secrets Bedside Cot Bed
Wonderful blog! I found it while browsing on Yahoo News. Do you have any
tips on how to get listed in Yahoo News? I’ve been trying for a
while but I never seem to get there! Appreciate it
What Robot Vac And Mop Experts Would Like You To Learn Robots That Vacuum And Mop
It’s great that you are getting ideas from this paragraph as well as from our dialogue
made here.
15 Top Pinterest Boards Of All Time About Buy A Driving License Without Advance Payment registrierten Führerschein Kaufen
10 Things That Your Family Taught You About Door Windows Replacement Door
Windows Replacement (Classicalmusicmp3Freedownload.Com)
Nine Things That Your Parent Teach You About Doors With Windows
Doors With Windows
Guide To Foldable Bedside Crib: The Intermediate Guide In Foldable Bedside
Crib foldable bedside crib (Lyda)
This is a topic that is close to my heart… Take care!
Exactly where are your contact details though?
Best Car Locksmith Near Luton Techniques To Simplify Your Daily Life Best Car Locksmith Near Luton Technique Every Person Needs
To Learn Best car locksmith near luton
The 10 Scariest Things About Best Auto Locksmith Luton auto Locksmith luton
Three Reasons Why Your Best Car Locksmith Luton Is Broken (And How To Repair It) Broken
The Unknown Benefits Of Auto Locksmiths Luton best auto locksmiths near luton (Lazaro)
Best Car Locksmith In Luton: What’s No One Has Discussed
Broken
Guide To Best Car Locksmiths In Luton: The Intermediate Guide For Best
Car Locksmiths In Luton car locksmiths in luton (Alexis)
Luton Car Locksmiths Tips From The Most Effective
In The Business Luton auto Locksmith
The 10 Most Terrifying Things About Best Car Locksmith In Luton best Car locksmith in luton
7 Helpful Tips To Make The Profits Of Your Car Locksmith Luton near
You’ll Never Be Able To Figure Out This Best Car Locksmiths
Luton’s Tricks best car Locksmiths luton
9 Things Your Parents Taught You About Best Auto Locksmiths Luton auto Locksmiths luton
What Luton Car Locksmith Could Be Your Next Big Obsession? auto locksmith in luton – tongcheng.jingjincloud.cn,
See What Car Locksmith Luton Tricks The Celebs Are
Utilizing car locksmith luton (Wilburn)
The 9 Things Your Parents Teach You About Best Auto Locksmiths In Luton best
auto Locksmiths in Luton (http://www.metooo.es)
You’ll Never Be Able To Figure Out This Auto Locksmiths Luton’s Tricks Auto Locksmiths Luton
10 Things Everybody Has To Say About Car Locksmiths Luton Car Locksmiths
Luton luton Car locksmiths
See What Auto Locksmiths Luton Tricks The Celebs Are Utilizing
auto locksmiths luton (Mildred)
See What Auto Locksmiths Near Luton Tricks The Celebs Are Making Use Of
auto locksmiths near luton
Why Luton Car Locksmith Is Harder Than You Think best Luton Car Locksmith
You’ll Never Be Able To Figure Out This Best Car Locksmiths Luton’s Tricks best car locksmiths luton – Lavon –
Why We Our Love For Auto Locksmith Luton (And You Should Too!) Best Luton auto Locksmith
Buy Cayden Yorkshire 101: Your Ultimate Guide For Beginners Axel Terrier Welpen Kaufen
– Bezauberndeyorkiewelpen58363.Blogdemls.Com –
See What Auto Locksmiths Near Luton Tricks
The Celebs Are Utilizing Auto locksmiths near Luton
20 Myths About Beds Bunk Beds: Dispelled Bunk beds for kids
If you want to get much from this paragraph then you have to apply these strategies to your won web site.
It’s The Myths And Facts Behind Pragmatic Slots Experience 프라그마틱 정품확인방법
Why Do So Many People Are Attracted To Buy The IMT Driving License?
Comprar Carta de ConduçãO da Categoria B1
The Reasons To Focus On The Improvement Of Bonnie Scarlet Macaw For Sale
Buy Macaw, Ngan,
There Is No Doubt That You Require Buy Arvin Biewer Yorkshire Terrier Mini Axel terrier
welpen kaufen (https://sciencewiki.science/wiki/10_Things_We_All_Love_About_Buy_Bruno_Mini_Yorkshire_Terrier)
10 Meetups About Buy Cayden Yorkshire You Should Attend arvin biewer Yorkshire terrier mini Kaufen
Spot on with this write-up, I really believe this website needs a lot more attention. I’ll probably be returning to
see more, thanks for the information!
Its like you read my mind! You appear to know so much about this,
like you wrote the book in it or something. I think that you could do with some pics to drive the message home a bit, but other than that, this is great
blog. A great read. I’ll certainly be back.
While it can be taken without food, it’s far better soaked up when taken with food.
My web site … https://Vnseo.Edu.vn/members/comiccoke41.293763.html
See What Baby African Grey Parrot For Sale Tricks The Celebs Are Making Use Of baby african grey parrot for sale – Amie –
11 Ways To Completely Revamp Your Britta Yorkshire Terrier Puppies For Sale Duxi yorkshire biewer kaufen (Michaela)
The Little-Known Benefits Of Buy Category B1 Driving License Comprar carta de condução da categoria C
Be On The Lookout For: How Buy Category B1 Driving License Is Taking Over
And What You Can Do About It Comprar Carta de condução categoria B –
pharmavam.ru –
14 Smart Ways To Spend Your Left-Over Buy Category B Driving License Budget Comprar Carta
de Condução da Categoria B1 (Sean)
See What Top Exercise Bikes Tricks The Celebs Are Using
exercise bikes
See What Vacuum Mop Cleaner Robot Tricks The Celebs Are Using vacuum mop cleaner robot
10 Life Lessons That We Can Learn From Sinatra Macaw For Sale Near Me Price Of Blue Macaw
See What Alex The African Grey Parrot Tricks The Celebs Are Utilizing Alex the african grey parrot (https://global.gooseket.com/member/Login.html?returnurl=https://charmingafricangreyparrotforsale.com/)
Responsible For An Harlequin Macaw Budget? Twelve Top Ways To Spend Your Money types of macaw
10 Reasons You’ll Need To Be Aware Of Britta Yorkshire Terrier
Puppies For Sale bruno mini yorkshire terrier Kaufen
Hyacinth Macaw Parrots For Sale: 11 Thing That You’re Failing To Do severe macaw price
15 Funny People Working In Buy The IMT Driving License In Buy The
IMT Driving License Comprar a carta De condução do imt
I always spent my half an hour to read this webpage’s
articles everyday along with a mug of coffee.
I am in fact thankful to the owner of this web page who has
shared this fantastic paragraph at here.
7 Things You’ve Never Known About Pallet Wood For Sale
pallets near me; Kisha,
20 Misconceptions About Buy Category A Driving License:
Busted comprar carta de condução da Categoria b1
How Luton Best Car Locksmith Was The Most Talked About Trend In 2024 best auto locksmiths in luton (Patrice)
Test: How Much Do You Know About Buy Category A Driving License?
Comprar carta de condução da categoria C
15 Reasons To Not Overlook Auto Locksmith In Luton car
I was skeptical at initially, however ProDentim surpassed my assumptions. My teeth are whiter, and my gum tissues are healthier. The natural, non-GMO components provide me satisfaction. ProDentim is currently a staple in my day-to-day oral care regimen.
My blog – http://Istiqbolsari.uz/user/chefparcel7/
Ziggy Our Scarlet Macaw Tools To Streamline Your Daily Lifethe One Ziggy
Our Scarlet Macaw Trick Every Individual Should Be
Able To Price of blue macaw
9 Lessons Your Parents Taught You About High-Quality
Factory-Made Pallets High-Quality Factory-Made Pallets (http://Www.517Yst.Com)
Here’s A Little-Known Fact About Auto Locksmith Luton best luton car locksmiths
5 Killer Quora Answers To Pram Newborn pram Newborn
9 Lessons Your Parents Teach You About Double Car Seat
Stroller double car seat stroller, taksim.in,
Since starting Nagano Lean Body Tonic, I have actually noticed significant enhancements in my digestion and energy degrees. The green tea extract and turmeric job marvels. It’s a natural and effective way to sustain weight management. I’m extremely pleased with the outcomes and will certainly proceed using it.
My web site: http://Www.Drugoffice.Gov.hk/gb/unigb/pattern-wiki.win/wiki/Natural_Methods_to_Weight_Monitoring_with_Lean_Body_Tonics
Do Not Buy Into These “Trends” Concerning Double Stroller side by
side double stroller (Deb)
A Intermediate Guide Towards Address Collection 주소모음사이트 (https://ucgp.jujuy.Edu.ar/)
The 9 Things Your Parents Taught You About Window Pane Replacement Window pane replacement
Best Car Locksmith Near Luton Tools To Streamline Your Daily Life Best Car Locksmith Near Luton Trick That Everybody Should Learn best car Locksmith near Luton
The 10 Most Scariest Things About Best Auto Locksmith Luton Best Auto Locksmith Luton
7 Little Changes That’ll Make A Huge Difference In Your Mental Health Private Care mental health online assessment uk (Alice)
Car Locksmith Luton Tools To Facilitate Your Day-To-Day Life Best Car locksmith Luton
How To Beat Your Boss With Best Car Locksmith In Luton best luton Car locksmith
VisiSoothe has actually been a superb enhancement to my everyday regimen. As a person who spends hours in front of a computer system, my eyes were continuously exhausted and strained. Since beginning VisiSoothe, I’ve observed a significant reduction in eye tiredness and enhanced clarity. It’s a natural, reliable option for keeping eye wellness. I can’t think of returning to life without it!
Feel free to visit my webpage: https://fcc.gov/fcc-bin/bye?https://cameradb.review/wiki/Improve_Your_Vision_Naturally_with_VisiSoothe
See What Gas Heating Engineer Near Me Tricks The Celebs Are
Making Use Of gas heating engineer near me (Lindsay)
Are You Responsible For An Leather Recliner Budget?
10 Ways To Waste Your Money Leather Grey Recliner
15 Best Documentaries About Car Locksmith Near Luton best luton car locksmith
See What Auto Locksmiths Near Luton Tricks The Celebs
Are Using auto locksmiths near luton
5 Killer Quora Answers On Auto Locksmith In Luton auto locksmith in luton (https://kingranks.com)
9 Lessons Your Parents Teach You About Best Auto Locksmiths
Luton best auto Locksmiths luton
10 Auto Locksmiths Near Luton Tips All Experts Recommend best luton Auto Locksmiths
15 Best Auto Locksmith In Luton Bloggers You
Should Follow best auto Locksmith in luton
The 10 Most Terrifying Things About Key Car Replacement key car replacement
The Reasons To Focus On Enhancing Chestnut Fronted Macaw how much do blue macaws cost (Leif)
The 12 Worst Types Of The Twitter Accounts That You Follow
주소모음
Best Car Locksmith In Luton It’s Not As Hard As You Think Auto locksmith in luton
20 Rising Stars To Watch In The Car Locksmiths Luton Industry Luton Car Locksmith
Guide To Best Car Locksmiths In Luton: The Intermediate Guide For Best Car Locksmiths In Luton car Locksmiths in Luton
You’ll Be Unable To Guess Car Locksmith
In Luton’s Secrets Car Locksmith In Luton
What’s The Current Job Market For Best Auto Locksmith Near Luton Professionals?
best auto locksmith near luton (Novella)
5 Car Locksmith Luton Projects For Every Budget best car locksmith luton – Elvin –
Think You’re Ready To Start Car Locksmiths Luton? Take This Quiz luton car locksmiths (Deanna)
9 Lessons Your Parents Teach You About Best Auto
Locksmiths Luton Best Auto Locksmiths Luton
Three Greatest Moments In Auto Locksmith Luton History auto locksmith in luton –
Hyman –
Guide To Car Locksmith Near Luton: The Intermediate Guide For Car Locksmith
Near Luton Car locksmith near luton
What Freud Can Teach Us About Best Car Locksmith Luton best luton Car locksmiths
The Greatest Sources Of Inspiration Of Luton Best Car Locksmith
best Auto locksmiths in luton
Five Killer Quora Answers To Best Car Locksmith
Luton Best car locksmith luton
10 Wrong Answers To Common Luton Auto Locksmith Questions Do You Know The
Right Answers? auto locksmith in luton
Item Upgrading Techniques To Simplify Your Daily Lifethe One Item Upgrading
Trick That Should Be Used By Everyone Learn Item Upgrading
The 10 Most Scariest Things About Car Locksmiths Luton Car Locksmiths Luton
5 Killer Quora Answers To Car Locksmith In Luton car Locksmith in luton
The Best Address Collection Site The Gurus Have Been Doing 3 Things 주고모음 (http://www.longisland.Com)
Ten Link Collection Sites That Really Improve Your Life
주소머음
5 Killer Quora Answers On Auto Locksmiths Near Luton auto locksmiths near luton
Pallet Near Me Tools To Help You Manage Your Everyday Lifethe Only Pallet Near Me Trick That Every
Person Must Know pallet near me
The 10 Most Terrifying Things About Pedal Exerciser Pedal Exerciser
5 Killer Quora Answers On Northern Containers northern containers
Five Buy A German Driving License Lessons From
The Pros deutscher führerschein zu verkaufen (Teena)
This paragraph will help the internet people for building up new blog or even a weblog from start to end.
How Buy A Driving License Rose To The #1 Trend In Social Media Deutscher FüHrerschein Zu Verkaufen
10 Websites To Help You Become An Expert In Hyacinth Macaw Parrots For Sale catalina macaw for sale;
Aisha,
Five Killer Quora Answers To Situs Alternatif Gotogel situs alternatif gotogel (Gonzalo)
What’s The Reason Everyone Is Talking About Pallet Wood For Sale Right Now Pallet Prices, 39.100.229.204,
Where Will Bluetooth Sex Machines One Year From Now?
sex Thrust machine
See What Auto Key Repair Near Me Tricks The Celebs Are Making Use Of auto
key repair; Scot,
Collection Of Site Addresses Explained In Fewer Than 140 Characters 주소모음 (Harrison)
What The Heck What Exactly Is Bonnie Scarlet Macaw For Sale?
Chestnut fronted macaw price (Bbs.zhizhuyx.com)
The Top Collection Of Addresses Gurus Are Doing Three Things 주소모움
Are You Responsible For An Address Collection Budget?
12 Top Ways To Spend Your Money 주소주라, Isaac,
How Repair Upvc Windows Arose To Be The Top Trend In Social Media upvc
windows locks, Everette,
Why Nobody Cares About Used Pallets For Sale where to get pallets (Fatima)
The 10 Most Scariest Things About Purchase Wood Pallets Purchase Wood pallets (kingranks.com)
Why Purchase Wood Pallets Still Matters In 2024 pallets near me (Holly)
14 Smart Ways To Spend Your Extra Money Link Daftar Gotogel Budget situs alternatif gotogel, https://crackwash3.werite.net/,
How To Outsmart Your Boss On Buy Marta Mini Yorkshire Terrier bruno mini
yorkshire Terrier kaufen (king-wifi.win)
It’s A Item Upgrade Success Story You’ll Never Be Able To
item level Upgrade
How To Tell The Replacement Sealed Units That’s Right For You Replacement sealed units for double glazed windows
3 Reasons Your Buy A2 Motorcycle License Online Is Broken (And How To Repair It) a2 motorradführerschein online Bestellen
You’ll Never Guess This New Wood Pallet For Sale’s Benefits New Wood Pallet For Sale
15 Top Documentaries About Power Tools Store power tools shop online – Leigh,
The Most Effective Advice You’ll Receive About Belgian Shepherd Dog Puppies
For Sale Austria deutscher schäferhund lebenserwartung
– servergit.itb.Edu.ec,
The Reasons You Should Experience Damian The Puppy At The Very Least Once In Your Lifetime Bruno Mini Yorkshire Terrier Kaufen (https://Bitroid.Ru/Bitrix/Redirect.Php?Goto=Https://Bezauberndeyorkiewelpen.De/)
The Little-Known Benefits Of Electric Treadmill Machine Basic Electric Treadmill
The Myths And Facts Behind Purchase Used Pallets Large Pallets
I just like the valuable info you provide for your articles.
I’ll bookmark your blog and check again here regularly. I am slightly certain I’ll learn many
new stuff right right here! Good luck for the next!
Beware Of This Common Mistake With Your Upvc Door Handles Patio Door Handle
9 Lessons Your Parents Teach You About Best Mid Sleeper Cabin Bed Best Mid Sleeper Cabin Bed
Unquestionably believe that which you said. Your favorite reason appeared to be on the web the simplest thing to be aware of.
I say to you, I certainly get irked while people think about worries that they plainly do not know about.
You managed to hit the nail upon the top and also defined out the whole thing without having side effect , people can take a signal.
Will probably be back to get more. Thanks
Repair Upvc Window Explained In Fewer Than 140 Characters seals
10 Misconceptions Your Boss Holds Concerning Buy A Polish Driving License kup prawo jazdy kat b+ (Antoine)
5 Killer Quora Answers On Heavy Duty Bariatric Wheelchair
bariatric wheelchair (Kelle)
The Benefits Of Buy Driving License Darknet At Least Once In Your Lifetime deutschen führerschein online kaufen (Thanh)
20 Tips To Help You Be More Efficient At The Swedish Traffic Agency’s Opening Hours förnya körkort högsbo
5 Laws Everybody In Buy A Polish Driving License Should Be
Aware Of kup prawo jazdy b (https://Kupprawojazdyonline71736.blazingblog.com/)
15 Top Pinterest Boards From All Time About Repairing Upvc
Windows upvc Window Locks
Thanks for sharing such a pleasant thinking, post is fastidious, thats
why i have read it entirely
10 Things You Learned In Kindergarden They’ll Help
You Understand ADHD And Anxiety Medication treat adhd without Medication Adults
A New Trend In German Shepherd Protective Instinct
german Shepherd puppies
10 Unexpected Buy Category B1 Driving License
Tips Comprar a carta de condução do IMT
The Top 5 Reasons Why People Are Successful With The Buy
Driving License Darknet Industry echten führerschein Kaufen
A Help Guide To Upvc Windows And Doors Near Me From Beginning To End Wickes Upvc Windows
3 Reasons You’re Not Getting Melody Blue Spix Macaw Isn’t Working (And The Best Ways To
Fix It) catalina Macaw for sale
What’s The Current Job Market For Bike Exercise Home Professionals?
bike exercise home (git.Fuwafuwa.moe)
The No. 1 Question Everybody Working In Keys Repair Should Be Able To Answer emergency car key repair (Christie)
Wow, this article is pleasant, my younger sister is analyzing these kinds of things,
therefore I am going to tell her.
See What Car Locksmith Near Bedford Tricks The Celebs
Are Utilizing car locksmith Near bedford
You’ll Never Guess This Car Locksmiths Near Luton’s Secrets car locksmiths near
luton (Dedra)
How To Tell If You’re Ready For Buy A Driving License kup prawo jazdy online – Stanton –
10 Quick Tips About Buy Axel Terrier Puppies Cayden Yorkshire Kaufen (Muse.Union.Edu)
Everything You Need To Know About Window Repair Near window repair near me
You’ll Never Guess This Sash Window Repair’s Tricks Sash Window
The Most Profound Problems In Best Car Locksmith High Wycombe Best Car Locksmith In High Wycombe
10 Misconceptions That Your Boss May Have Regarding Keys Repair car key repair
How Do You Explain Adhd Private Assessment To A 5-Year-Old adhd assessment psychiatry uk
The Most Common Mistakes People Make With Replacement Lost Car Keys lost car keys Replacement cost uk
7 Helpful Tips To Make The Profits Of Your Goethe Certificate B1 Goethe-Zertifikat B1 online kaufen
You’ll Never Guess This Buy Pallets Near Me’s Tricks Buy Pallets Near Me
15 Funny People Working In Buy The IMT Driving License In Buy
The IMT Driving License Comprar Carta De ConduçãO Categoria B
3 Ways The Best Car Locksmiths Luton Can Affect Your Life car
locksmiths in luton (Loretta)
10 Things You Learned In Preschool That’ll Help You With Leather Recliners leather couch Recliners
The 10 Scariest Things About Best Auto Locksmith
Luton auto locksmith luton
See What Luton Car Locksmiths Tricks The Celebs Are Utilizing luton car locksmiths
See What Luton Car Locksmiths Tricks The Celebs Are Using luton car
locksmiths (Raymundo)
Why Do So Many People Would Like To Learn More About Auto Locksmith Near Luton? Auto Locksmith Luton
Why Windows Repair Could Be More Dangerous
Than You Realized installation
15 Gifts For The Tallula Indigo Park Mollie Macaw Lover In Your Life Catalina
macaw for sale (http://0lq70ey8yz1b.com/home.php?Mod=space&uid=903641)
Guide To African Grey Parrot Adoption: The Intermediate Guide On African Grey Parrot Adoption african grey parrot adoption (Penney)
Guide To Good Exercise Bicycle: The Intermediate Guide On Good Exercise Bicycle good exercise bicycle;
Owen,
15 Secretly Funny People Work In Auto Locksmiths In Luton Best luton car Locksmiths
Car Locksmiths Luton: It’s Not As Difficult As You Think best car locksmiths in luton (http://www.metooo.co.uk)
20 Irrefutable Myths About Car Locksmith Near Luton: Busted nearby
A Journey Back In Time: What People Discussed About Buy Category B Driving License 20 Years Ago Comprar carta de Condução da categoria C
The Most Profound Problems In Buy Category B Driving
License comprar a carta de condução do imt
10 Meetups Around Folding Treadmills You Should Attend Treadmills fold up
How Buy French Bulldog Puppies Has Become The Most Sought-After Trend In 2024 bulldogge Kaufen
Why You’ll Need To Find Out More About Auto Locksmith Luton replacements
Don’t Buy Into These “Trends” About Buy The IMT Driving License Comprar Carta de Condução da Categoria B1
Buy Axel Terrier Puppies Tools To Streamline Your Daily Life Buy Axel
Terrier Puppies Trick Every Individual Should Know Axel Terrier Welpen kaufen
The People Nearest To Auto Locksmith Luton Have Big Secrets
To Share Nearest
Why Do So Many People Want To Know About Buy Category A Driving License?
Comprar Carta de Condução da Categoria B1
The Buckingham Heating Engineers Success Story You’ll Never Remember Gas Safe Registered Engineer Buckingham [Eric1819.Com]
What Is Buy Category B1 Driving License And Why Are We
Talking About It? Comprar carta de condução da categoria C,
Joesph,
How Buy Category B1 Driving License Became The Hottest Trend In 2024 Comprar carta de condução da categoria C
(https://www.tmbv68.ru/go.php?link=https://www.cartoexpressodeportugal-96b.com/)
This Story Behind Buy Category B1 Driving License Is One That Will Haunt You Forever!
Comprar Carta De ConduçãO Da Categoria B1
20 Up-And-Comers To Watch In The Buy The IMT
Driving License Industry comprar carta de condução Da categoria a (botdb.win)
What’s The Current Job Market For Best Auto Locksmith Near Luton Professionals?
best auto locksmith near luton (Rick)
See What Car Locksmith Luton Tricks The Celebs Are
Using Car Locksmith Luton – https://Morphomics.Science/Wiki/10_Facts_About_Luton_Best_Car_Locksmith_That_Will_Instantly_Get_You_Into_A_Great_Mood,
Are You Getting The Most Out Of Your Car Locksmiths In Luton? key
The Ugly Truth About Where To Buy Chest Freezer where
can i buy a small chest freezer near me (https://olderworkers.com.au/author/prxmz634ii6-sarahconner-Co-uk)
10 Things You Learned From Kindergarden They’ll Help You Understand Buy
Category B Driving License Comprar Carta de Condução da Categoria B1 (Vince)
How To Explain Treatment Of ADHD In Adults To Your
Grandparents best Online adhd treatment – Elearnportal.Science –
The Motive Behind Buy Category B1 Driving License Will Be
Everyone’s Desire In 2024 Comprar a carta de condução do IMT
The Three Greatest Moments In Buy Counterfeit Money History Hochwertige FäLschungen
How Much Do Ferrari Key Fob Replacement Uk Experts Earn? Ferrari Car Keys
The 10 Most Terrifying Things About Compact Mobility Scooters Compact Mobility scooters (mobility-Scooters34710.bcbloggers.Com)
15 Reasons To Not Ignore Hamlin Candle Arch French Bulldog kaufen Oder adoptieren
Ten Honda Keys That Really Change Your Life Honda keys (https://minecraftcommand.science/profile/rootlibra66)
Why Buy The IMT Driving License Could Be Your Next Big Obsession? Comprar carta De condução categoria B (elearnportal.science)
The 10 Most Terrifying Things About Driving Lessons Scunthorpe driving lessons scunthorpe
Battery Power Tool Set: 10 Things I’d Loved To Know Earlier battery
power tool sets (Carole)
The Comprehensive Guide To Buy Bruno Mini Yorkshire Terrier
Marta Mini Yorkshire Terrier Kaufen
Why Buy A1 Driving License Online Is The Right
Choice For You? köPa svenskt körkort
A Time-Travelling Journey: How People Talked About Buy Arvin Biewer
Yorkshire Terrier Mini 20 Years Ago Cayden yorkshire kaufen
12 Companies That Are Leading The Way In Buy The IMT Driving License Comprar A Carta de condução do IMT
It’s Time To Expand Your Hob Options Ovens And Hobs Uk (http://Www.Hulkshare.Com)
15 Amazing Facts About Britta Yorkshire Terrier Puppies For Sale Cayden yorkshire kaufen;
Essie,
10 Things Everyone Hates About Paisley Hyacinth Macaw For Sale
Paisley Hyacinth Macaw For Sale severe Macaw Price
Buy Marta Mini Yorkshire Terrier: What No One Is Talking About Mini Biewer Yorkshire Terrier Kaufen
15 Buffy Macaw Bird For Sale Benefits That Everyone Should Be
Able To macaw Shop
7 Things About Purebred German Shepherd Breeder You’ll Kick
Yourself For Not Knowing deutscher schäferhund welpe kaufen (Caleb)
5 Laws That’ll Help The Robot Vacuum Industry robot
vacuum Deals (Pattern-wiki.win)
5 Things That Everyone Doesn’t Know On The Subject Of Assessment In Mental Health Mental Health Assessment Report
Why Ziggy Our Scarlet Macaw Is Relevant 2024 Severe Macaw Price
Why You’ll Want To Learn More About Mindy Catalina Macaw spix macaw lifespan (https://www.bioguiden.se)
Tips For Explaining Best Robot Vacuums To Your Boss automatic hoover robot,
Tandy,
10 Easy Ways To Figure The Leather Recliners For Sale You’re Looking For cheap couches for sale under $100 [Garry]
This blog was… how do I say it? Relevant!! Finally I have found something which helped me.
Cheers!
How Much Can Great Green Macaw Experts Make? buy macaw parrot (Harley)
How To Create Successful Stationary Cycle For Exercise
Techniques From Home Spinning Bike
20 Insightful Quotes On German Shepherd Protective
Instinct German Shepherd Puppies
How Buy German Shepherd Dog Switzerland Can Be Your Next Big
Obsession deutsche schäferhunde kaufen (Bella)
10 Easy Steps To Start The Business Of Your Dream Sinatra Macaws For
Sale Business Chestnut-fronted Macaw price (m.yonginseoulbone.com)
The 3 Most Significant Disasters In Private Psychiatrist History how Much do Private psychiatrists Charge
What’s The Current Job Market For Talking African Grey
Parrot For Sale Professionals? african grey parrot for sale (bbs.zhizhuyx.com)
10 Untreated ADHD In Adults Uk Hacks All Experts Recommend severe untreated adhd in adults
7 Small Changes That Will Make A Huge Difference In Your Bonnie Scarlet Macaw For Sale Hyacinth Macaw Cost
The 10 Most Scariest Things About Buy Macaw Buy Macaw (Billy)
How To Explain Ghost Immobiliser Fitting Birmingham To Your Grandparents
ghost immobiliser reviews (Heike)
Why Buy German Shepherd Dog Switzerland Still Matters In 2024 German Shepherd Kaufen
15 Up-And-Coming Buy A French Bulldog Bloggers You Need To See FranzöSische Bulldogge Kaufen Hamburg
See What Macaw Keycaps Tricks The Celebs Are Using macaw keycaps (Maxwell)
10 Hythian Macaw For Sale That Are Unexpected
macaw pet shop
5 Laws Anyone Working In Buy Category B1 Driving License Should Know comprar carta de condução da categoria B1 – https://intern.ee.aeust.edu.tw/,
The Most Hilarious Complaints We’ve Heard About Buy A Real Driver’s License Acheter Un Permis De Conduire EuropéEn, Articlescad.Com,
The Most Significant Issue With Exercise Bicycle And
How To Fix It exercise bicycles For Sale
10 Apps That Can Help You Manage Your Buy Category B Driving License
Comprar carta de condução categoria B
Are Buy The IMT Driving License As Important As Everyone Says?
Comprar Carta de Condução da Categoria B1 (Darnell)
A Journey Back In Time What People Said About Buy Category B Driving License 20 Years Ago comprar carta de condução da categoria a
10 Inspiring Images About Dewalt Tool Set Power Tools Dewalt
13 Things You Should Know About Buy Category C Driving License That You Might Not Have Known comprar a carta
de condução do imt (leo-ug.ru)
Five Lessons You Can Learn From Buy A German Shepherd deutscher schäferhund
pflege (Clyde)
This Is The History Of Buy Category C Driving License In 10 Milestones Comprar Carta De ConduçãO Da Categoria A (Cameradb.Review)
5 Common Myths About Buy Category B1 Driving License You
Should Stay Clear Of Comprar carta de condução da categoria C
Ten Buy Category A Driving License-Related Stumbling Blocks You Should
Not Share On Twitter Comprar a carta de Condução do IMT
15 Gifts For The Orville Macaw Parrot Price Lover
In Your Life do macaws make good pets
How To Identify The Right Buy Category B1 Driving License For You Comprar
Carta De ConduçãO Categoria B (https://Restoprep-Ideas.My-Free.Website)
The History Of Buy Wheel Loader Driving License Online In 10 Milestones
körkort i cv (Penney)
10 Things We All Were Hate About Key Fob Repairs key
fob repairs near me (Kristina)
The 15 Things Your Boss Would Like You To Know You’d Known About Buy Macaw macaw Pet for sale
15 Pinterest Boards That Are The Best Of All Time About ADHD
Without Medication treating adhd without medication [Tomas]
Link Daftar Gotogel Tips To Relax Your Everyday Lifethe Only Link Daftar Gotogel Trick
That Everybody Should Learn link Daftar gotogel
The 3 Most Significant Disasters In Chestnut Fronted Macaw History spix macaw characteristics – Fred,
See What Situs Gotogel Terpercaya Tricks The Celebs Are Utilizing Situs Gotogel Terpercaya, Hikvisiondb.Webcam,
A Delightful Rant About Severe Macaw spix macaw for Sale (Bagjury7.bravejournal.net)
The Most Successful Macaw Purchase Experts Have Been Doing 3 Things macaw price –
Ezequiel,
One Of The Biggest Mistakes That People Make With Tallula
Indigo Park Mollie Macaw macaw birds to buy
You’ll Be Unable To Guess Exterior Door With Window’s Benefits Exterior Door With Window [http://Git.Bkdo.Net]
Five Killer Quora Answers On Window Door Company
window door company
Buy A Driving License Legally: What’s The Only Thing Nobody
Is Talking About FüHrerschein Zum Kaufen
Are You Confident About Doing Private Psychiatrist In London? Take This Quiz private psychological evaluation
What’s The Job Market For Website Gotogel Alternatif Professionals?
website Gotogel alternatif (https://Idvideo.site/)
Guide To Situs Gotogel: The Intermediate Guide On Situs
Gotogel situs gotogel (Margart)
The 10 Scariest Things About Double Glazed Door Handles double glazed Door handles
How To Create An Awesome Instagram Video About Buy German Shepherd Baby deutscher schäferhund Schwarz kaufen (http://www.Survivalmonkey.Com)
10 Things That Your Family Teach You About Recommended Cribs recommended
Cribs (89.234.183.97)
The 12 Worst Types Of Users You Follow On Twitter upvc windows and doors (Randal)
See What Adult ADHD Psychiatrist Near Me Tricks The Celebs
Are Making Use Of Adhd psychiatrist near me (yogicentral.Science)
5 Laws That Can Help The German Shepherd To Give Away Industry schäFerhund welpe Kaufen
Buy German Shepherds The Process Isn’t As Hard As You
Think deutsche SchäFerhundwelpen
The 10 Most Scariest Things About Situs Gotogel situs gotogel (https://able2Know.org/user/flavorperch2/)
школа хип хопа взлом [url=https://apk-smart.com/igry/dlya-devochek/909-shkola-hip-hopa-vzlom.html]https://apk-smart.com/igry/dlya-devochek/909-shkola-hip-hopa-vzlom.html[/url] школа хип хопа взлом
P.S Live ID: K89Io9blWX1UfZWv3ajv
P.S.S [url=http://mss-k.com/bbs/board.php?bo_table=in&wr_id=4028]Программы и игры дл[/url] [url=https://www.livejournal.com/login.bml?returnto=http%3A%2F%2Fwww.livejournal.com%2Fupdate.bml&event=%D0%B2%D0%B7%D0%BB%D0%BE%D0%BC%20demolition%20derby%20%5Burl%3Dhttps%3A%2F%2Fapk-smart.com%2Figry%2Fgonki%2F261-demolition-derby-crash-racing-vzlomannaja-mnogo-deneg.html%5Dhttps%3A%2F%2Fapk-smart.com%2Figry%2Fgonki%2F261-demolition-derby-crash-racing-vzlomannaja-mnogo-deneg.html%5B%2Furl%5D%20%D0%B2%D0%B7%D0%BB%D0%BE%D0%BC%20demolition%20derby%20%0D%0A%20%0D%0AP.S%20Live%20ID%3A%20K89Io9blWX1UfZWv3ajv%20%0D%0AP.S.S%20%5Burl%3Dhttps%3A%2F%2Fglitteronlights.com%2Fpendant-lights%2F%5D%D0%9F%D1%80%D0%BE%D0%B3%D1%80%D0%B0%D0%BC%D0%BC%D1%8B%20%D0%B8%20%D0%B8%D0%B3%D1%80%D1%8B%20%D0%B4%D0%BB%D1%8F%20%D0%90%D0%BD%D0%B4%D1%80%D0%BE%D0%B8%D0%B4%20%D1%82%D0%B5%D0%BB%D0%B5%D1%84%D0%BE%D0%BD%D0%B0%5B%2Furl%5D%20%5Burl%3Dhttp%3A%2F%2Fstophish.ru%2Fnews%2Fget%2F5129%5D%D0%9F%D1%80%D0%BE%D0%B3%D1%80%D0%B0%D0%BC%D0%BC%D1%8B%20%D0%B8%20%D0%B8%D0%B3%D1%80%D1%8B%20%D0%B4%D0%BB%D1%8F%20%D0%90%D0%BD%D0%B4%D1%80%D0%BE%D0%B8%D0%B4%20%D1%82%D0%B5%D0%BB%D0%B5%D1%84%D0%BE%D0%BD%D0%B0%5B%2Furl%5D%20%5Burl%3Dhttps%3A%2F%2Fwww.livejournal.com%2Flogin.bml%3Freturnto%3Dhttp%253A%252F%252Fwww.livejournal.com%252Fupdate.bml%26event%3Dflash%2520on%2520call%2520%25D1%2581%25D0%25BA%25D0%25B0%25D1%2587%25D0%25B0%25D1%2582%25D1%258C%2520%253Ca%2520href%253Dhttps%253A%252F%252Fapk-smart.com%252Fprogrammy%252F376-flash-on-call-polnaja-versija.html%253Ehttps%253A%252F%252Fapk-smart.com%252Fprogrammy%252F376-flash-on-call-polnaja-versija.html%253C%252Fa%253E%2520flash%2520on%2520call%2520%25D1%2581%25D0%25BA%25D0%25B0%25D1%2587%25D0%25B0%25D1%2582%25D1%258C%2520%250D%250A%2520%250D%250AP.S%2520Live%2520ID%253A%2520K89Io9blWX1UfZWv3ajv%2520%250D%250AP.S.S%2520%25202c4065e%2520%5D%D0%9F%D1%80%D0%BE%D0%B3%D1%80%D0%B0%D0%BC%D0%BC%D1%8B%20%D0%B8%20%D0%B8%D0%B3%D1%80%D1%8B%20%D0%B4%D0%BB%D1%8F%20%D0%90%D0%BD%D0%B4%D1%80%D0%BE%D0%B8%D0%B4%20%D1%82%D0%B5%D0%BB%D0%B5%D1%84%D0%BE%D0%BD%D0%B0%5B%2Furl%5D%20%20aeaf8c6%20]Программы и игры для Андроид телефона[/url] [url=http://probki.kirov.ru/content/reshenie-20112013-0#comment-31733322]Программы и игры для Андроид телефона[/url] 41c27ce
Five Killer Quora Answers On Alternatif Gotogel
Terpercaya alternatif gotogel terpercaya (sheehan-mccall-2.federatedjournals.Com)
See What Buy A Purebred German Shepherd Tricks The Celebs Are Making Use Of
Buy a purebred German Shepherd
The 10 Most Scariest Things About Togel
4d togel 4d (skovgaard-Vance-2.blogbright.net)
The Little-Known Benefits Of Gotogel situs gotogel Terpercaya – kupicabel.ru,
What’s The Current Job Market For Psychiatrists ADHD Near Me
Professionals Like? Psychiatrists adhd near Me
Beware Of These “Trends” Concerning Private Mental Health Online Mental Health Assessment
10 Things That Your Family Taught You About Adhd
Assessments For Adults adhd assessments for adults
A Provocative Remark About Gotogel Link Alternatif bocoran Togel
20 Fun Facts About Buy Fakes Hochwertige fälschungen
Guide To Togel 4d: The Intermediate Guide To Togel 4d
togel 4D
What’s The Job Market For Gotogel Professionals Like?
gotogel
5 Robot Vacuum Lessons Learned From Professionals Robotic vacuum cleaner
14 Smart Ways To Spend On Leftover ADD Treatments For Adults Budget treatment for adhd and ptsd combined – Julianne,
A Productive Rant About Composite Door Crack Repair composite Door repair
See What Link Daftar Gotogel Tricks The Celebs Are Utilizing Link Daftar Gotogel
How To Get Better Results From Your Buy German Shepherd
Reinrassiger Deutscher SchäFerhund Kaufen
24 Hours To Improving Melody Blue Spix Macaw mini macaw For sale
(gdeotveti.ru)
7 Tricks To Help Make The Most Of Your Buy Bismarck Yorkshire Terrier Puppies suzie Der yorkie-welpe
Situs Alternatif Gotogel Tools To Ease Your Daily Lifethe One Situs Alternatif Gotogel
Trick That Every Person Should Be Able To situs alternatif gotogel
20 Questions You Need To To Ask About Buy Mini Biewer Yorkshire Terrier Prior
To Purchasing Buy Mini Biewer Yorkshire Terrier Duxi
yorkshire biewer kaufen (Bezaubernde-yorkie-welpen62346.Azzablog.com)
Need Inspiration? Check Out French Doors And Side Windows french doors with side Windows
Comprehensive List Of Bi Fold Door Repair Dos And
Don’ts bi fold door repairs (Nidia)
5 Killer Quora Answers To Bi Fold Door Repairs Near Me Bi fold door repairs near me
(https://king-wifi.win/)
Enough Already! 15 Things About Vauxhall Insignia Key Replacement We’re Sick Of Hearing replacement car key fob vauxhall
There’s A Reason Why The Most Common Fridgemaster Chest Freezer Debate It’s
Not As Black And White As You Might Think chest freezers reviews Uk
15 Funny People Working In Adult ADHD Test In Adult ADHD Test Adhd Assessment Test For Adults [Intern.Ee.Aeust.Edu.Tw]
What’s The Current Job Market For Exterior Doors And Windows Professionals Like?
Exterior doors And Windows
10 Reasons Why People Hate Buy Bruno Mini Yorkshire Terrier.
Buy Bruno Mini Yorkshire Terrier Cayden yorkshire kaufen
15 Trends That Are Coming Up About Blue Shepherds deutsche Schäferhunde Welpen Kaufen österreich
The Ugly The Truth About Private Mental Healthcare mental health assessment Uk
The One ADHD Tests Trick Every Person Should Know Adhd Test For adults (valetinowiki.racing)
The Most Inspirational Sources Of Link Alternatif Gotogel alternatif gotogel terpercaya, Tania,
You’ll Be Unable To Guess Link Alternatif Gotogel’s
Secrets link alternatif Gotogel (coopers-chicago96582.Imblogs.Net)
Are You Sick Of Trustworthy Counterfeit
Money Sellers? 10 Inspirational Resources To Rekindle Your Love 100% echtes falschgeld
3 Ways That The Buy Driving License A1 Influences Your Life kup prawo jazdy a1 online (https://Pattern-wiki.win/Wiki/11_Creative_Ways_To_Write_About_Buy_A_Driving_License)
See What Toto Macau Tricks The Celebs Are Using Toto macau
Toto Macau Tips To Relax Your Daily Life Toto Macau Trick
That Every Person Should Learn toto macau (Margarito)
What A Weekly Adult ADHD Psychiatrist Near Me Project Can Change Your Life Private Practice Psychiatry Near Me, http://Wx.Abcvote.Cn/Home.Php?Mod=Space&Uid=4219182,
The Most Convincing Proof That You Need Order A2
Driving License Online a2 motorradführerschein online
erwerben – Evie –
What’s The Most Creative Thing Happening With Buy Driving License deutschen führerschein kaufen
Ten Easy Steps To Launch The Business Of Your Dream ADHD In Adult Women Business Diagnosing adhd In women
Ten Situations In Which You’ll Want To Be
Aware Of ADHD Medication Adhd Medication Online
10 Quick Tips For ADHD In Women inattentive adhd in adult women
Situs Alternatif Gotogel Techniques To Simplify Your Everyday
Lifethe Only Situs Alternatif Gotogel Trick That Should Be Used By Everyone Learn situs Alternatif gotogel
Why Nobody Cares About ADHD Test Adult online adhd tests – Deloras,
See What Maxi Cosi Rotating Car Seat Tricks The Celebs Are Utilizing maxi cosi rotating car seat
Remarkable things here. I am very satisfied to see your post.
Thanks so much and I’m looking forward to touch you.
Will you please drop me a e-mail?
The 9 Things Your Parents Teach You About Link Daftar Gotogel
link daftar gotogel
Forget Buy A Motorcycle Driving License A1 And A2: 10
Reasons That You No Longer Need It registrierten führerschein kaufen – https://qooh.me/
–
20 Myths About Titration ADHD Medications: Busted titration for adhd – http://www.google.com.om,
Why Bentley Smart Key Still Matters In 2023 bentley activation Key
Guide To Situs Gotogel: The Intermediate Guide
Towards Situs Gotogel situs gotogel
20 Trailblazers Leading The Way In Gotogel alternatif gotogel terpercaya
How To Make A Successful Buy Driving License Tips From Home köp Sverige körkort
The Most Underrated Companies To Keep An Eye On In The
Front Doors With Windows Industry Companies
Guide To Symptoms Of ADHD In Adult Women: The Intermediate Guide
To Symptoms Of ADHD In Adult Women Symptoms of adhd In adult women
5 Clarifications On Togel 4d Game Judi Togel
This Week’s Most Popular Stories About Psychiatrist Private can A private psychiatrist prescribe medication
The 10 Most Scariest Things About Link Login Gotogel
link login gotogel (Louise)
Guide To Alternatif Gotogel Terpercaya: The
Intermediate Guide On Alternatif Gotogel Terpercaya Alternatif gotogel terpercaya
A Provocative Remark About Buy A Driving License füHrerschein kaufen seriöS
The 10 Most Terrifying Things About Composite
Door Repair Near Me composite door repair near me (k12.instructure.com)
You’ll Be Unable To Guess ADHD Stimulant Medication’s Tricks Adhd stimulant medication (120.zsluoping.cn)
9 Things Your Parents Teach You About ADHD Assessment
Adults Uk Adhd Assessment Adults Uk
You’ll Never Guess This Psychiatry ADHD Near Me’s Tricks Psychiatry Adhd Near me
Situs Alternatif Gotogel Tools To Ease Your Daily Lifethe One Situs Alternatif Gotogel Technique Every
Person Needs To Learn situs alternatif gotogel (Theresa)
Will Mental Health Clinic Be The Next Supreme Ruler Of The World?
Mental Health Assessment Online
Five Killer Quora Answers On Bandar Togel Terpercaya bandar togel terpercaya (findmeacaregiver.com)
A Productive Rant About Buy A Driving License Legally
registrierten führerschein kaufen
10 Situs Togel Terpercaya Strategies All The Experts Recommend
10 situs togel terpercaya (Marcia)
See What Built In Microwave Black Tricks The Celebs Are Using built in microwave black
(http://www.v0795.com)
10 Reasons You’ll Need To Know About Psychiatrist Uk Private Private psychiatrist Hitchin
9 Signs That You’re An Expert Buy Bismarck Yorkshire Terrier Puppies Expert Suzie Der Yorkie-Welpe
It’s The Complete Cheat Sheet On Private Psychiatrist
Birmingham private psychiatric assessment near me [e-directory2u.com]
Buy A German Shepherd 101″The Ultimate Guide For Beginners deutscher Schäferhund Welpe kaufen
The Advanced Guide To Space Key Lamborghini key lamborghini aventador
– Randell –
10 Situs Togel Terpercaya Strategies All The Experts Recommend 10 situs togel terpercaya (Lynn)
Why Do So Many People Want To Know About Auto Locksmith Near Buckinghamshire?
locksmiths
The 10 Most Terrifying Things About Sale Cot sale cot (https://efkdc.ru/redirect?url=https://www.Cots4Tots.Co.uk)
You’ll Be Unable To Guess Link Alternatif Gotogel’s Secrets link Alternatif Gotogel (http://120.zsluoping.cn)
The Top Reasons Why People Succeed In The ADHD
Assessment Private Industry Private Adhd assessments
10 Tips For Quickly Getting Audi Car Key Replacement replacement Key for Audi A4
This Is A Bandar Togel Terpercaya Success Story You’ll Never Be Able To Situs Paling Terpercaya Togel
Are Website Gotogel Alternatif As Vital As Everyone
Says? rumus Togel
Why People Are Talking About Ziggy Our Scarlet Macaw Today blue
macaw price (Grady)
A Good Rant About What Is A Ghost Immobiliser
Immobiliser Ghost
What’s The Current Job Market For 10 Situs Togel Terpercaya Professionals Like?
situs togel terpercaya; https://hikvisiondb.webcam/wiki/A_StepBy_Step_Guide_For_Choosing_Your_Bandar_Togel_Terpercaya,
5 Killer Quora Answers To French Windows And Doors french windows
and doors, Therese,
You’ll Be Unable To Guess Gotogel Link Alternatif’s Benefits gotogel
It is perfect time to make some plans for the longer term and it is time to be happy. I have read this submit and if I could I want to recommend you few attention-grabbing issues or advice. Perhaps you can write subsequent articles referring to this article. I wish to read more issues approximately it!
Feel free to visit my site – https://lafeng666.com/home.php?mod=space&uid=174713
You’ll Be Unable To Guess Link Alternatif Gotogel’s Tricks link alternatif gotogel
(https://imoodle.Win)
This Is The German Driving License For Sale
Case Study You’ll Never Forget kann man einen führerschein kaufen, qna.lrmer.com,
The No. Question Everybody Working In Samsung Side By Side Fridge Freezer Uk Should Know How To Answer fridge price samsung (Olga)
Situs Alternatif Gotogel Tools To Streamline
Your Everyday Lifethe Only Situs Alternatif Gotogel Trick That Everybody Should Learn situs Alternatif gotogel
What Is The Buy The IMT Driving License Term And How To Utilize It Comprar A Carta De ConduçãO Do IMT
See What Bariatric Folding Wheelchair Tricks The Celebs Are Utilizing bariatric
folding wheelchair (Rickie)
The Most Significant Issue With Psychiatrist Assessment, And How You Can Repair
It psychiatry uk adhd self assessment (Sabrina)
All The Details Of Buy The IMT Driving License
Dos And Don’ts Comprar carta de Condução da categoria A
10 Failing Answers To Common Buying A Driving License Experience Questions: Do You Know The Right Ones?
FüHrerschein Kaufen Darknet
5 Things Everyone Gets Wrong On The Subject Of Test For ADHD
In Adults adhd symptoms test
One Mercedes Replacement Key Cost Success Story You’ll Never Believe key for mercedes (Tory)
Buy Category B1 Driving License Tips From The Best In The Industry Comprar Carta de Condução da Categoria B1
5 Killer Quora Answers On Situs Gotogel Terpercaya Situs Gotogel terpercaya
Signs And Symptoms Of ADHD In Adults Tools To Ease Your Everyday Lifethe Only Signs
And Symptoms Of ADHD In Adults Trick That Every Person Should
Learn signs and symptoms Of adhd in adults
10 Things You Learned In Kindergarden That’ll Help You
With Upvc Windows Repair Upvc Window Repairs Near Me
A Comprehensive Guide To Refrigerators Samsung From Start To Finish samsung Brand Refrigerator
Ten Myths About Swedish Driving License Online That Don’t Always Hold Köpa C Körkort Online (Beatrice)
It’s genuinely very difficult in this full of activity life to listen news on TV, so I just
use internet for that reason, and obtain the newest news.
The 10 Scariest Things About Gotogel Link Alternatif gotogel link alternatif – https://jbels.net/x/cdn/?https://gotogelreal.lol,
The Unknown Benefits Of Psychiatrists Near Me private pay psychiatrist near me
(Margo)
What Is Robot Vacuum And How To Use It Robotic Vacuum Cleaner
10 Private Psychiatrist Belfast Cost Tricks All Pros Recommend
how much is a private psychiatrist (Malinda)
20 Resources That Will Make You Better At Replacement Windows
Bristol Door Repair Bristol
You’ll Never Guess This 40ft Tunnel Containers’s Benefits 40ft tunnel container [Josie]
The 10 Most Scariest Things About Toto Macau toto Macau
The 10 Most Scariest Things About Top Fridge Freezer Brands which fridge is the best (Ramon)
See What Auto Key Repair Near Me Tricks The Celebs Are Using Auto Key Repair
Are You Getting The Most You Bmw Car Key Replacement? keyless
entry bmw (Ledbookmark.Com)
Do Not Make This Blunder With Your Buy Arvin Biewer Yorkshire Terrier Mini Bruno Mini Yorkshire Terrier Kaufen
You’ll Never Guess This Private Psychiatrist Near Me’s Benefits Private psychiatrist near me
The Top 5 Reasons People Thrive In The Mini Cycle Exercise Bike Industry Leg exercise machine
How To Create An Awesome Instagram Video About Buy Category A Driving License Comprar carta de condução Da categoria A
What NOT To Do With The Buy A German Shepherd Industry Deutscher SchäFerhund Pflege
Replacement Lock For Composite Door Tips To Relax Your Daily Lifethe One
Replacement Lock For Composite Door Trick That Everybody Should Be Able To replacement lock for composite Door
20 Things That Only The Most Devoted Mazda Dealership Key Replacement Fans Understand Mazda Key replacement cost (http://forexmob.ru/user/silkatom56/)
Tallula Indigo Park Mollie Macaw: It’s Not As Difficult As You Think
Macaw Bird Toys
You’ll Never Be Able To Figure Out This Situs Alternatif Gotogel’s Secrets Situs Alternatif Gotogel
See What Add ADHD Medications Tricks The Celebs Are Making Use Of Add Adhd Medications
The Worst Advice We’ve Heard About Buy A Real Driver’s License échange permis
De conduire France (jcbbscn.com)
The French Bulldog For Sale Puppies Case Study You’ll Never Forget kaufen oder adoptieren (Imogene)
Window & Door Tools To Make Your Daily Life Window & Door Trick That Should Be Used By Everyone Learn window & door (smed-fischer-4.technetbloggers.de)
Guide To Cost Of Private ADHD Assessment UK: The
Intermediate Guide On Cost Of Private ADHD Assessment UK cost
of private adhd assessment uk (https://minecraftcommand.science/profile/capcost44)
The Best Place To Research Gotogel Online togel Online
10 Tell-Tale Symptoms You Need To Get A New Buy Mini Biewer Yorkshire Terrier
Bruno mini yorkshire terrier kaufen
Situs Gotogel Terpercaya Tools To Streamline Your Daily Life Situs Gotogel Terpercaya Technique Every Person Needs To Know situs gotogel (https://2ch-ranking.net/redirect.php?url=https://telegra.ph/20-Myths-About-Situs-Alternatif-Gotogel-Dispelled-02-06)
Assessments For ADHD In Adults Tools To Streamline Your Everyday Lifethe Only
Assessments For ADHD In Adults Trick Every
Individual Should Know assessments for adhd in adults
10 Things Your Competitors Teach You About Upvc Windows Repair home
10 Facts About Doors Windows Uk That Will Instantly Put You In A Good Mood Casement
See What Audi Replacement Key Service Near Me Tricks The Celebs Are Using audi replacement key service near Me
What Is Best Freezer Brands And Why Is Everyone Talking About It?
coolest fridges
For Whom Is Buy A Legal Driving License And Why You Should Take
A Look registrierten führerschein kaufen ohne
anzahlung (Irwin)
The 10 Most Terrifying Things About Keys Mercedes Keys Mercedes
How To Explain Buy Mini Biewer Yorkshire Terrier To Your Grandparents Marta Mini Yorkshire Terrier Kaufen
The 10 Most Scariest Things About Titration Meaning ADHD titration meaning Adhd (molchanovonews.ru)
You’ll Be Unable To Guess Ferrari Key Programming Near Me’s Benefits Near me
20 Quotes Of Wisdom About Buy Category A Driving License Comprar carta de condução da categoria C
Your Worst Nightmare About Togel 4d It’s Coming To Life Togel Deposit Pulsa
See What Baby African Grey Parrot For Sale Tricks The Celebs Are Using Baby african grey parrot for sale
Watch Out: How Emergency Patio Door Repair Is Taking Over And What You Can Do About It service
Five Killer Quora Answers On Repair Hole In Composite Door repair hole in composite door
The 10 Most Terrifying Things About Hahns Macaw For Sale hahns macaw for sale
You’ll Be Unable To Guess Situs Gotogel’s Benefits
Situs Gotogel
The 10 Scariest Things About Legit Crypto Casino legit crypto casino;
freebookmarkstore.win,
Guide To Alternatif Gotogel Terpercaya: The Intermediate Guide In Alternatif Gotogel Terpercaya alternatif gotogel terpercaya (lt.dananxun.cn)
The Three Greatest Moments In Mental Health Assessment Uk History mental health assessor (Marcy)
What’s The Point Of Nobody Caring About Buy Category C Driving License Comprar Carta de Condução da Categoria B1
Five Killer Quora Answers To Website Gotogel
Alternatif gotogel (cruisaway.bmgroup.be)
15 Best Documentaries On Buy Clovis Yorkshire Terrier Duxi yorkshire biewer kaufen – https://naturalbookmarks.com –
10 Things That Your Family Taught You About Remote Car Key Repair remote car key Repair (http://shenasname.ir/)
10 Things That Your Family Taught You About ADHD In Adults Symptoms Women adhd in Adults symptoms Women
How To Make An Amazing Instagram Video About Adult Add Women women and attention deficit disorder (Carmella)
What’s The Current Job Market For Car Locksmith Bedford Professionals Like?
Car locksmith bedford (https://fakenews.win/)
20 Things You Need To Be Educated About Private Psychiatrist Liverpool Cost private psychiatric assessment london (https://eurosynapses.Giannistriantafyllou.gr/)
A Look At The Ugly Reality About Buy Registered Driving License Online deutscher eu führerschein kaufen
Why All The Fuss Over Replacement Audi Key? audi spare key (Winifred)
This Week’s Top Stories Concerning Gotogel Situs gotogel Terpercaya
10 Real Reasons People Hate Purebred German Shepherd Breeder reinrassiger deutscher Schäferhund züchter
15 Fun And Wacky Hobbies That’ll Make You More Successful At Buy Clovis Yorkshire Terrier Bruno mini yorkshire terrier kaufen
5 Clarifications On Buy Arvin Biewer Yorkshire Terrier Mini Arvin biewer yorkshire terrier mini kaufen
Guide To Situs Togel Resmi: The Intermediate Guide The Steps To Situs Togel
Resmi situs Togel resmi
10 Things That Everyone Is Misinformed Concerning Skoda Key skoda octavia key programming
Five Things You Didn’t Know About Argos Fridge Freezer fridge freezer deals (https://tv-detail.ru/bitrix/click.php?anything=here&goto=https://www.frydge.uk)
How To Survive Your Boss On Buy A Driving License fuhrerschein-kaufen-ohne-vorkasse
This Week’s Top Stories Concerning Buying A German Driving License
Experiences Fuhrerschein Kaufen
What’s The Job Market For Cheap Samsung Fridge Freezers
Professionals? cheap samsung fridge freezer (Elisha)
See What Situs Alternatif Gotogel Tricks The
Celebs Are Making Use Of Situs Alternatif Gotogel
Could Private Diagnosis For ADHD Be The Key To Dealing With 2023?
private adhd assessment north east (Kendall)
14 Businesses Doing A Great Job At Doors With Windows French doors with windows
10 Meetups About Adhd Private Assessment You Should
Attend get adhd assessment – https://www.google.pt/url?q=https://yogicentral.science/wiki/15_reasons_You_must_love_affordable_adhd_assessment –
Buzzwords De-Buzzed: 10 Other Methods To Say Buy
Category A Driving License Comprar Carta de Condução Da Categoria B1
A Comprehensive Guide To Mental Assessment. Ultimate Guide To Mental
Assessment mental health assessment for court; https://morphomics.science/wiki/This_Is_The_Ultimate_Guide_To_Mental_Health_Specialist_Near_Me,
5 Killer Quora Answers To In Built Microwave Oven In built Microwave oven
See What Situs Togel Terbesar Tricks The Celebs Are Using
situs Togel terbesar
5 Killer Quora Answers On L Shape Bunk Bed l shape Bunk
You’ll Never Guess This Gotogel Link Alternatif’s Benefits Gotogel Link Alternatif
10 Top Mobile Apps For Kia Sportage Replacement Key kia key replacement
near me (http://Www.stes.tyc.edu.tw/xoops/modules/profile/Userinfo.php?uid=1876161)
Why Do So Many People Are Attracted To Mindy Catalina Macaw?
Where To buy macaws
13 Things You Should Know About Buy Category B Driving License That You Might Not Know Comprar carta de
condução da categoria A (Katrin)
10 Fundamentals Regarding Upvc Doors Windows You Didn’t Learn At School fitters
How Do You Know If You’re Set To Go After Mini Cooper Replacement
Keys mini key fob not working
What To Do To Determine If You’re Prepared To Go After Buy Cayden Yorkshire Mini Biewer Yorkshire Terrier Kaufen
Situs Alternatif Gotogel Tools To Make Your Everyday Lifethe Only Situs
Alternatif Gotogel Trick Every Individual Should Be Able
To Situs Alternatif Gotogel
Guide To Ghost Immobiliser And Tracker: The Intermediate Guide On Ghost Immobiliser And Tracker ghost immobiliser and Tracker
What Is Car Locksmiths Near Milton Keynes And Why Is Everyone Dissing It?
Best Car Locksmiths Near Milton keynes
The Top Companies Not To Be In The Double Glazed Units Manufacturers Near Me Industry fitted
Nine Things That Your Parent Taught You About Lexus Ct200h Key Replacement Cost lexus ct200H key replacement cost
Hyacinth Macaw Parrots For Sale’s History History Of Hyacinth Macaw Parrots For
Sale Mini macaw Price (clashofcryptos.trade)
See What Link Alternatif Gotogel Tricks The Celebs Are Utilizing
Link alternatif Gotogel (gotogelreal78125.popup-blog.com)
“The Gotogel Awards: The Most Stunning, Funniest, And Weirdest Things We’ve Ever Seen Alternatif Gotogel terpercaya
8 Tips To Increase Your Hyundai I10 Key Game hyundai i10 key Fob replacement
14 Creative Ways To Spend Extra Link Daftar Gotogel Budget togel Sgp
What Is Order A2 Driver’s License Online And Why Is Everyone Speakin’ About It?
führerschein a2 online erwerben (Catalina)
Driving License Category C Explained In Fewer Than 140 Characters Prawo Jazdy b1
Why You’ll Definitely Want To Find Out More About Female Adult
Toys adult toys dildos
Consultant Psychiatrist Near Me Tools To Help You Manage Your Daily Life
Consultant Psychiatrist Near Me Trick Every Individual Should Learn Consultant psychiatrist near me
12 Companies Leading The Way In Buy A Driving
License legalen führerschein kaufen
Situs Alternatif Gotogel Tools To Improve Your Daily Life
Situs Alternatif Gotogel Trick That Everybody Should Learn Situs alternatif gotogel
Five Things You’ve Never Learned About Glassrepair residential glass window repair
15 Undeniable Reasons To Love Bentley Continental Key Fob
Prices
The 10 Most Scariest Things About Best Auto Locksmith
In Buckinghamshire best auto locksmith in buckinghamshire [freeman-mcculloch.mdwrite.net]
See What Website Gotogel Alternatif Tricks The Celebs Are Making Use Of website gotogel alternatif
20 Fun Infographics About German Shepherd Puppies For Sale
In Switzerland deutscher schäferhund kaufen schweiz
You’ll Be Unable To Guess Link Alternatif Gotogel’s Secrets link Alternatif Gotogel
20 Reasons Why Upvc Door Doctor Near Me Will Not Be Forgotten Tailored window repair
The 10 Most Terrifying Things About Link Daftar Gotogel link daftar
gotogel (Amee)
It Is The History Of ADHD In Adult Women Symptoms In 10
Milestones adhd in women symptoms (Deborah)
What’s The Job Market For Website Gotogel Alternatif Professionals Like?
website gotogel alternatif (Angelita)
The 3 Largest Disasters In Buying A German Driving License
Experiences History führerschein zum kaufen (Deana)
You’ll Never Guess This Rolls Royce Smart Key’s Benefits rolls royce smart key (Wanda)
See What Buy Real Driving License UK Tricks The Celebs Are Utilizing buy real driving license uk (Tilly)
Symptoms ADHD In Adults Tools To Improve Your Everyday Lifethe Only Symptoms
ADHD In Adults Trick That Everybody Should Learn symptoms adhd in adults
A Glimpse Into American Fridge’s Secrets Of American Fridge 80cm wide american fridge freezer (Marina)
The Ultimate Cheat Sheet On Volvo V50 Key volvo xc60 emergency key
What Window Handles You’ll Use As Your Next Big Obsession repair door
handle (https://www.metooo.Es)
What’s The Job Market For Gas Engineer Boiler Professionals?
gas engineer boiler – Pearl,
Avoid Making This Fatal Mistake You’re Using Your Fridge-Freezer cheap fridges uk (ddht.ru)
Since I started making use of Provadent, my oral examinations have actually been better. My teeth are stronger, and my gum tissues are healthier. The natural formula is gentle yet reliable. Provadent is currently a staple in my day-to-day routine.
Here is my web page http://www.Jzq5.cn/space-uid-356142.html
What’s The Current Job Market For Buy Category B Licence
Online Professionals? Buy Category B Licence Online
The Unspoken Secrets Of Skoda Car Key Replacement skoda key replacement
Stationary Bike Exercise: 10 Things I’d Like To Have Learned Sooner Portable Exercise Bike
Link Login Gotogel Techniques To Simplify Your Daily Life Link Login Gotogel Trick That Every
Person Should Be Able To link login gotogel
The Top Psychiatrist Near Me Tricks To Transform Your Life top Psychiatrist near me
What’s The Job Market For Lamborghini Car Keys Professionals Like?
lamborghini car Key
10 Of The Top Mobile Apps To Bonnie Scarlet Macaw For Sale Buy macaws
20 Things You Need To Be Educated About Buy Macaw Macaw Purchase
How To Choose The Right Online Mental Health Assessment
On The Internet mental health trauma assessment (Penney)
Why Macaw Purchase Is Your Next Big Obsession catalina macaw for sale (http://Www.nzdao.cn)
Five Replacement Key For Bentley Continental Gt Lessons From
The Pros Workshop
A Guide To Buy A Category B+ Driving License From Beginning To End kup
prawo jazdy b online (Bit1024.site)
20 Irrefutable Myths About Collection Of Site Addresses: Busted 주소 모음 (http://www.play56.Net)
20 Fun Details About German Shepherd Puppies schäferhundwelpen kaufen
14 Businesses Doing A Great Job At Adhd Assessments For Adults how do you get Assessed for adhd
What’s The Job Market For ADHD Assessment For Adults Professionals Like?
Adhd Assessment for Adults
The Little-Known Benefits Of Door Handle Replacement Front Door Handle Repair
14 Cartoons About Treatment Of ADHD In Adults To Brighten Your Day non medication treatment For Adhd Adults
Private Psychiatrist Nottingham Tools To Ease Your Daily Lifethe
One Private Psychiatrist Nottingham Trick That Everyone Should Learn psychiatrist – August –
Built-In Fridge Freezer’s History History Of Built-In Fridge Freezer Best integrated fridge freezer 70/30
20 Things You Need To Know About ADHD Private Diagnosis Adhd Diagnosis
Private Cost (Yanyiku.Cn)
It’s The Good And Bad About Private Psychiatrist Northern Ireland private Psychiatrist glasgow cost
This Week’s Top Stories About Doors With Windows Doors With Windows French doors with Windows
The Best Psychiatrist Near Me Tricks To Transform Your Life
best Psychiatrist near me (marvelvsdc.faith)
11 Methods To Completely Defeat Your Undiagnosed ADHD In Women adhd Screening for women
The 10 Most Scariest Things About Situs Alternatif Gotogel situs alternatif gotogel
I’ve been browsing online more than 2 hours today, yet I never found any interesting article like yours.
It is pretty worth enough for me. Personally, if all site owners and bloggers made
good content as you did, the internet will be a lot more
useful than ever before.
The Most Hilarious Complaints We’ve Heard About Upvc Window Repairs upvc window repairs near me [Brook]
Private ADHD Clinic Tips That Can Change Your Life Private adhd Assessment middlesbrough
How To Determine If You’re In The Right Place For Buy A Registered Driver’s License Without
A Deposit führerschein kaufen deutschland
15 Terms Everybody Who Works In Buy Bruno Mini Yorkshire Terrier
Industry Should Know Clovis yorkshire terrier kaufen (https://account.safecreative.org/checkSession?r=https://bezauberndeyorkiewelpen.de/)
Why All The Fuss? Citroen Replacement Key Cost? citroen ds3 car Key – https://minecraftcommand.science/profile/augustwave50,
10 Websites To Help You Develop Your Knowledge About Paisley Hyacinth Macaw For Sale catalina
macaw price (Sheldon)
See What Link Daftar Gotogel Tricks The Celebs Are Making Use Of Link Daftar gotogel (King-wifi.win)
25 Unexpected Facts About Adult ADD Treatments treatment for adhd and ptsd Combined
– https://Valetinowiki.racing
–
Best Car Locksmiths High Wycombe: A Simple Definition Best Car Locksmiths In High Wycombe
5 Must-Know Psychiatrists Near Me-Practices You Need To Know For 2024 private psychiatrist near me uk
10 Signs To Watch For To Know Before You Buy Situs Alternatif Gotogel
Toto Togel
Situs Gotogel Terpercaya Tools To Streamline Your Daily Lifethe One Situs Gotogel Terpercaya Technique Every
Person Needs To Learn situs gotogel
Nine Things That Your Parent Teach You About Black 3 Seater Fabric Sofa
black 3 seater fabric sofa
10 Tell-Tale Symptoms You Need To Find A New ADHD Diagnosis Adults late
adhd diagnosis (Astrid)
10 Simple Steps To Start Your Own Mercedes Keys Business mercedes car key (compravivienda.com)
Cost Of African Grey Parrot Tools To Ease Your Daily
Lifethe One Cost Of African Grey Parrot Trick That Every Person Must Be Able To cost of african grey parrot; Alton,
Cat Flap Fitters Near Me cat flap fitters (Tangela)
You Are Responsible For A Gotogel Budget? 12 Best Ways To Spend Your Money Link daftar Gotogel
Private Psychiatrist Online 101:”The Complete” Guide For Beginners private Psychiatrist Bedford
You Are Responsible For An Buy A German Shepherd Budget?
12 Tips On How To Spend Your Money blaue schäferhunde
(mgbg7b3bdcu.net)
20 Resources That’ll Make You More Efficient With Electric Folding Wheelchair bariatric foldable electric wheelchair (Kellie)
Guide To Gotogel Link Alternatif: The Intermediate Guide Towards Gotogel Link Alternatif gotogel link alternatif
Are You Getting The Most Of Your Buy A2 Motorcycle License Online?
FüHrerschein dauer
Why You Should Concentrate On The Improvement Of
Buy Category B Driving License Comprar a carta de condução do IMT
Ten Things You Learned In Kindergarden They’ll Help You Understand Buy Category B Driving License
Comprar Carta de Condução categoria B (http://testbusiness.tabgametest.de)
Why You Should Focus On Enhancing Buy The IMT Driving License Comprar Carta de Condução Da Categoria B1 [https://rewardlyre00.bravejournal.net]
I was recommended this website by my cousin. I am not sure whether this post is written by him as no
one else know such detailed about my difficulty.
You are wonderful! Thanks!
See What Add Adult Women Tricks The Celebs
Are Using add adult women
10 Misconceptions Your Boss Has About Damian The Puppy Suzie Der Yorkie-Welpe
Three Common Reasons Your ADHD Diagnosis UK Adults Isn’t Working (And How To Fix It) how do you Diagnose adhd In adults
Guide To Replacement Conservatory Windows: The Intermediate Guide In Replacement Conservatory Windows replacement conservatory windows – Eden
–
How Much Do Titration Process Experts Make?
adhd titration meaning (Kendra)
The 10 Most Scariest Things About Private Adhd Assessment
London Adhd Assessment London
Case Battle Tools To Help You Manage Your Daily Lifethe One Case Battle Trick That Every Person Must Be Able To csgo
case battles (Connie)
Door Handle Replacement Tips From The Best In The Business Double Glazing Door Handle Repairs
How To Create An Awesome Instagram Video About Buy A Driving License Mofaführerschein kaufen; bbs.airav.Cc,
The 10 Scariest Things About Situs Togel
Terbesar situs Togel terbesar, http://www.guzhen0552.Cn,
Nine Things That Your Parent Taught You About Best Car Locksmith
Near Milton Keynes best car locksmith near milton keynes (Hamish)
You’ll Never Be Able To Figure Out This Gas Fitters Milton Keynes’s
Tricks Gas Fitters Milton Keynes
Bmw Key: It’s Not As Difficult As You Think Bmw key replacement Service
See What Website Gotogel Alternatif Tricks The Celebs Are Utilizing website gotogel
alternatif (Anthony)
Its History Of Car Locksmith Bedford fob
Give Me The Address: The Good, The Bad, And The Ugly 주소링크모음
(https://lingkeumo-eumsaiteu88751.acidblog.net/62329991/20-Trailblazers-leading-the-way-in-address-collection-site)
Guide To Website Gotogel Alternatif: The Intermediate Guide On Website Gotogel Alternatif Website Gotogel Alternatif
10 ADHD Symptoms In Women Tricks Experts Recommend Signs Of Adhd Adult Women
Is Spare Key For Car The Most Effective Thing That Ever Was?
emergency car Key
ADHD Medication Adults Uk Tools To Ease Your Daily Lifethe One ADHD Medication Adults Uk Trick
Every Individual Should Know adhd medication adults uk (Samara)
What’s The Job Market For Replacement Bmw Key Professionals?
replacement bmw Key
How Buy A Driving License Changed Over Time Evolution Of Buy A Driving
License kupić Prawo jazdy bez stresu
10 Things We All Love About Seat Key Seat car key lost cost
See What Cost For Replacement Car Key Tricks The Celebs Are Using Cost for replacement car key
See What Situs Gotogel Terpercaya Tricks The Celebs Are Making Use Of Situs Gotogel terpercaya
See What Website Gotogel Alternatif Tricks The Celebs Are Using website gotogel alternatif
Five Qualities That People Search For In Every Buy Category A
Driving License Comprar carta de condução da categoria A – Geneva –
See What Wheelchair Ramp Folding Tricks The Celebs Are Making Use Of wheelchair ramp folding
Assessments For ADHD In Adults Tools To Ease Your Daily Life
Assessments For ADHD In Adults Trick Every Person Should Learn assessments for adhd in adults
20 Things You Need To Be Educated About Macaw Cage Macaw Pet
15 Best Exercise Cycles For Sale Bloggers You Need To Follow Commercial Exercise Bike
10 Unexpected Audi A3 Car Key Battery Tips audi a1
key replacement (Lorene)
5 Must-Know Practices For Private Psychiatrists In London In 2023
private psychological assessment [wwwiampsychiatrycom97158.ssnblog.Com]
24 Hours For Improving Glass Replacement Windows replacement glass for double glazing (Chanda)
What The 10 Most Worst ADHD Medication Ritalin Fails Of All
Time Could Have Been Prevented starting adhd Medication adults
10 Things That Your Family Taught You About Realisticsexdolls realisticsexdoll
What’s The Job Market For Anxiety Disorder Physical Symptoms Professionals?
anxiety disorder physical symptoms (Isiah)
15 Startling Facts About Car Locksmiths Near Milton Keynes That
You Didn’t Know About Best car locksmiths near milton keynes (gleason-gibson-3.federatedjournals.com)
9 Things Your Parents Teach You About Best Robot Hoover best robot hoover; http://monamiprofessional.com/bitrix/redirect.php?goto=https://www.robotvacuummops.uk,
What Is Mercedes Car Key Replacement And How To Utilize It mercedes benz key
15 Startling Facts About Buy Category C Driving
License You’ve Never Known Comprar carta de conduçãO da categoria A
See What Buy C1 E License Online Tricks The Celebs Are Making Use Of
Buy C1 E License Online (Maple)
Guide To Upvc Window Repair Near Me: The Intermediate Guide On Upvc Window Repair Near
Me Upvc window repair near me
20 Tips To Help You Be Better At Situs Gotogel situs gotogel terpercaya
(Julieta)
5 Common Myths About Land Rover Key You Should Avoid land rover discovery 3 replacement Key fob
It’s Time To Expand Your Website Gotogel Alternatif Options Situs Gotogel
Why Nobody Cares About ADHD Private Diagnosis London How Much Does Private Adhd Assessment Cost
The 10 Most Scariest Things About Gotogel Link Alternatif gotogel link
alternatif (Vince)
You’ll Never Be Able To Figure Out This Conservatory Door Repairs Near Me’s Benefits conservatory door repairs near me
10 Mistaken Answers To Common Gotogel Questions: Do You Know The Right Answers?
Togel terlengkap
How To Solve Issues Related To Compact Travel Scooters travel mobility Scooters
How To Beat Your Boss With 20 Ft Tunnel Container
20ft tunnel container – iblog.iup.edu –
How To Outsmart Your Boss On Buy German Shepherds Reinrassiger Deutscher SchäFerhund Welpe
It’s The Complete Cheat Sheet On Best Brand Fridge Freezer brands of fridge freezers
Consultant Psychiatrist Near Me Tools To Make Your Daily Lifethe One Consultant Psychiatrist Near Me
Technique Every Person Needs To Be Able To consultant psychiatrist near me
You’ll Never Guess This Private Consultant Psychiatrist’s Tricks Private consultant psychiatrist
Is Buffy Macaw Bird For Sale Really As Vital As Everyone Says?
Buy Macaw Bird
How You Can Use A Weekly Replace Window Handle Project Can Change Your Life door handle lock Repair [https://sixn.Net]
Speak “Yes” To These 5 Locksmiths Cars Tips carkey Locksmith
Buy The IMT Driving License Isn’t As Tough As You Think Comprar carta de condução categoria B (http://www.northwestu.edu)
How To Create An Awesome Instagram Video About Buy An Old German Shepherd Dog schäferhundwelpen kaufen (Lillie)
Where Are You Going To Find Britta Yorkshire Terrier Puppies For Sale Be
One Year From In The Near Future? cayden Yorkshire kaufen
Why Adding Gotogel To Your Life Can Make All The Impact Situs Gotogel
Titration ADHD Tools To Streamline Your Everyday Lifethe Only Titration ADHD Trick That Everyone
Should Learn titration adhd, Heriberto,
The Top Samsung American Style Fridge The Gurus Have Been Doing Three Things
samsung fridge freezer black (Stefanie)
The 10 Most Scariest Things About Realisticsex Doll realisticsex Doll
The 10 Most Terrifying Things About Rollator Walker With Seat Uk
rollator walker with seat uk
5 Reasons To Be An Online Damian The Puppy And 5 Reasons To
Not suzie der yorkie-welpe (caiman.ru)
See What Replacement Window Seals Tricks The Celebs Are Making Use Of Replacement Window Seals
See What Situs Alternatif Gotogel Tricks The Celebs Are Using situs alternatif gotogel
Hi Dear, are you truly visiting this website on a regular basis,
if so then you will absolutely get fastidious experience.
10 Mobile Apps That Are The Best For Buy Category C Driving License comprar carta De conduçãO da categoria B1
10 Inspirational Graphics About Buy A Driving License Without An Exam Deutschen führerschein Legal Kaufen
5 Laws That Will Help The Where Can I Get A Honda Key Cut
Industry honda civic replacement Key fob
5 Killer Quora Answers On Repair Bifold Door Top Roller repair bifold door top Roller
(https://nativ.media:443/wiki/index.php?pasteash95)
What’s The Current Job Market For ADHD In Adults Self Assessment Professionals Like?
adhd in adults self assessment (Bret)
What’s The Job Market For Pragmatic Homepage Professionals Like?
프라그마틱 무료체험 메타
5 Killer Quora Answers On Situs Gotogel gotogel
What The 10 Most Stupid Inattentive ADHD Medication Fails Of
All Time Could Have Been Avoided over the Counter adhd medication for adults
See What Situs Toto Tricks The Celebs Are Using situs toto
Ten Stereotypes About Upvc Door Locks That Don’t Always Hold replacing a upvc door lock
See What Composite Door Lock Replacement Tricks The Celebs Are Utilizing composite door lock replacement
The Most Underrated Companies To In The Buy Arvin Biewer Yorkshire Terrier Mini Industry
Bismarck Welpen Yorkshire Terrier Kaufen (Library.Hbs.Edu)
See What Situs Togel Terbesar Tricks The Celebs Are Using situs togel terbesar
Buy A German Shepherd Tips From The Most Effective In The Business Deutscher Schäferhund pflege
10 Websites To Help You To Become An Expert In Lovesense Machine sexmachines.co.Uk
“The Psychiatric Assessment UK Awards: The Most Stunning, Funniest, And Most Bizarre Things We’ve Seen how to get a psychiatric
assessment uk
What’s The Current Job Market For German Shepherd For Sale Professionals Like?
Deutscher SchäFerhund Kaufen Schweiz (https://Wiki.R4L.Com/Api.Php?Action=Https://Entzckendescferhundwelpen-Wec35D.De)
ADHD Medication Adults Uk Tools To Help You Manage Your Daily
Lifethe One ADHD Medication Adults Uk Trick That Every Person Should
Learn adhd medication
The Most Popular French Door Fridge Gurus Are Doing 3 Things French Door Fridge Vs Side By Side
15 Reasons Why You Shouldn’t Ignore Damian The Puppy Bismarck Welpen Yorkshire Terrier Kaufen
Guide To Situs Togel Resmi: The Intermediate Guide In Situs Togel Resmi situs togel Resmi
10 Situs Togel Terpercaya Tips All Experts Recommend 10 Situs togel Terpercaya
Learn What Buy A Purebred German Shepherd Tricks The Celebs Are Using
belgischer schäferhund welpen kaufen österreich
Are You Getting The Most You Diagnosis Of ADHD? Adhd Diagnosis London
The 10 Most Scariest Things About Windows And Doors Aluminium windows
and doors Aluminium (https://trade-britanica.trade/)
3 Reasons Commonly Cited For Why Your Purebred German Shepherd Dog Isn’t Performing (And Solutions To
Resolve It) einen deutschen schäferhund kaufen, Salvatore,
10 Wrong Answers To Common Treating Adult ADHD Questions: Do You Know The Right Ones?
treatment for severe adhd in adults (metooo.co.uk)
Do Not Buy Into These “Trends” About Toto Macau Tempat Bermain Togel
Guide To Situs Togel Terpercaya: The Intermediate Guide On Situs Togel Terpercaya Situs Togel Terpercaya
Five Killer Quora Answers To Repair Key Fob Repair Key
How To Survive Your Boss With Buy Marta Mini Yorkshire Terrier
axel terrier welpen kaufen
The 10 Most Terrifying Things About Auto Locksmith Near Northamptonshire
auto locksmith Near northamptonshire
What’s The Current Job Market For ADHD In Women Adults Professionals?
adhd in women adult
What’s The Job Market For Low Frost Fridge Freezers Professionals Like?
frost fridge freezer (Eva)
This History Behind Buy A German Driving License
Will Haunt You For The Rest Of Your Life! deutschland für den kauf eines Führerscheins
The 10 Most Scariest Things About ADHD Test For Women adhd test For women
Five Things You’ve Never Learned About Adult Treatment For ADHD what Can untreated adhd lead to
Buy The IMT Driving License’s History History Of Buy The IMT Driving License Comprar Carta de Condução da Categoria B1 (Bernie)
This Is The Advanced Guide To Buy Driving License
Online Pkw FüHrerschein Kaufen
Pragmatic Official Website: It’s Not As Expensive As You Think 프라그마틱 공식홈페이지; Rentry.co,
11 Methods To Totally Defeat Your Couches On Sale Corner Sofa
Sale (Spotlessmusic.Com)
Are You Responsible For A Pragmatic Free Budget? 10 Ways
To Waste Your Money 프라그마틱 불법
You’ll Never Be Able To Figure Out This Buy Pallets Near Me’s Benefits buy pallets Near me
Responsible For An Door Windows Replacement Budget?
12 Ways To Spend Your Money door window Replacement (https://lovewiki.faith/wiki/Tuckermcintyre5908)
Unexpected Business Strategies For Business That Aided Non Stimulant ADHD Medication Succeed adhd Medication over the counter
uk (tongcheng.jingjincloud.Cn)
It’s amazing in favor of me to have a website, which is
valuable in favor of my knowledge. thanks admin
15 Gifts For The Psychiatrist Near Me Private Lover In Your Life psychiatrists near (Sylvia)
10 Things That Your Family Taught You About Website Gotogel Alternatif Website gotogel Alternatif
You’ll Never Be Able To Figure Out This Best Auto Locksmith Near Milton Keynes’s Benefits Auto
Locksmith Near milton keynes (championsleage.review)
Think You’re The Perfect Candidate For Doing ADHD Symptoms Adult?
Do This Test adhd symptoms Symptoms
5 Killer Quora Answers On ADHD In Women Checklist Adhd In Women Checklist [http://Www.Google.St]
The 10 Most Scariest Things About Situs Gotogel Situs Gotogel
Link Daftar Gotogel Tools To Streamline Your Everyday Lifethe Only Link Daftar Gotogel Trick
Every Person Should Know link daftar gotogel; Moises,
Guide To Car Locksmiths Bedford: The Intermediate
Guide For Car Locksmiths Bedford car locksmiths bedford – Nicki –
Guide To Locksmith Near Me For House: The Intermediate Guide The Steps To Locksmith Near Me For House locksmith near me for house
The Leading Reasons Why People Achieve In The Adult Test For ADHD Industry where to get adhd testing (Christiane)
5 People You Oughta Know In The Buy Pallets UK Industry large pallets (Wayne)
You’ll Never Be Able To Figure Out This Add Symptoms In Adult Women’s Benefits add
symptoms in adult women – blogfreely.net –
The Story Behind Double Glaze Repair Near Me Can Haunt You
Forever! Double Glazed Windows Replacement
Upvc Windows Near Me Tools To Ease Your Daily Life Upvc
Windows Near Me Technique Every Person Needs To Be Able To upvc windows near me – ns1.javset.net,
20 Things You Need To Be Educated About Cars Keys Replacement lost car key
replacement near me; Genesis,
11 “Faux Pas” Which Are Actually OK To Create Using Your Buy German Shepherds schäferhundwelpen (https://andersson-pilgaard-2.mdwrite.net/the-reasons-why-buy-A-german-shepherd-has-become-everyones-obsession-in-2024/)
Buy Counterfeit Money Discreetly: A Simple Definition deutsche banknoten fäLschen
Why Landrover Keys Should Be Your Next Big Obsession land rover Leather Key case
You’ll Never Guess This ADHD Traits In Women’s Benefits types of
adhd in women (Claudia)
17 Reasons Why You Shouldn’t Ignore Bentley Car Keys bentley key repair (Vivien)
Hey! I know this is kinda off topic however , I’d figured I’d ask. Would you be interested in exchanging links or maybe guest authoring a blog article or vice-versa? My website covers a lot of the same topics as yours and I think we could greatly benefit from each other. If you are interested feel free to shoot me an email. I look forward to hearing from you! Great blog by the way!
my webpage; https://elearnportal.science/wiki/Healthy_And_Balanced_Consuming_Practices_for_Lasting_Weight_Loss
I have actually been utilizing VisiSoothe for a couple of months now, and the outcomes are extraordinary! My vision has significantly boosted, and I no much longer experience the eye pressure I used to. The natural active ingredients make me feel great regarding what I’m taking into my body. VisiSoothe is a game-changer for any individual aiming to enhance their eye health normally. Extremely advise!
Here is my webpage; https://www.webwiki.fr/birtheditor5.bravejournal.net/understanding-the-conveniences-of-a-vision-clearness-supplement
excellent points altogether, you just gained a new reader.
What would you suggest in regards to your publish that you made a few days in the past?
Any positive?
Incredible! This blog looks exactly like
my old one! It’s on a totally different subject
but it has pretty much the same layout and design.
Great choice of colors!
I wish to show my respect for your kindness in support of those people who must have help on your situation. Your personal commitment to passing the solution throughout had become exceedingly beneficial and has regularly allowed professionals just like me to reach their objectives. Your useful instruction entails a lot to me and far more to my peers. With thanks; from each one of us.
Feel free to surf to my web-site :: http://demo.Emshost.com/space-uid-2694434.html
I love reading through an article that will make
people think. Also, many thanks for permitting me to comment!
I’m not sure why but this site is loading incredibly slow for me.
Is anyone else having this problem or is it a
issue on my end? I’ll check back later and see if the
problem still exists.
Can you tell us more about this? I’d love to find out
some additional information.
Good day! This post couldn’t be written any better! Reading this post reminds me of my previous room
mate! He always kept talking about this. I will forward this article to him.
Fairly certain he will have a good read. Thank you for sharing!
VisiSoothe has been a great enhancement to my everyday regimen. As someone that invests hours in front of a computer, my eyes were regularly exhausted and stretched. Because beginning VisiSoothe, I’ve discovered a considerable decrease in eye fatigue and boosted clarity. It’s an all-natural, effective solution for maintaining eye health. I can not picture returning to life without it!
Also visit my site: https://nemoserver.Iict.bas.bg/hoseanull71660/jorja2005/wiki/Simple-Way-Of-Living-Modifications-for-Improved-Vision-Assistance
Thanks a lot for sharing this with all people you actually realize what you are speaking
about! Bookmarked. Please also consult with my website =).
We could have a link alternate contract between us
I was skeptical regarding vision supplements, however VisiSoothe has surpassed my assumptions. My vision has actually come to be more clear, and I feel much less pressure after lengthy days at the office. The blend of natural components is remarkable, and I value the focus on eye health and wellness. VisiSoothe is a must-try for any individual experiencing vision issues or wishing to maintain healthy and balanced eyes.
Feel free to surf to my web-site https://Rrallytv.com/@charissaritz13?page=about
I was hesitant regarding vision supplements, yet VisiSoothe has actually surpassed my expectations. My vision has actually become more clear, and I really feel much less stress after long days at the workplace. The mix of natural ingredients goes over, and I value the concentrate on eye health and wellness. VisiSoothe is a must-try for anybody experiencing vision concerns or wishing to maintain healthy eyes.
Also visit my webpage :: http://Pandora.Nla.Gov.au/external.html?link=https://anotepad.com/notes/hdktysx3
Appreciate you sharing, great blog article.Really looking forward to read more. Will read on…
Good day! This post couldn’t be written any better! Reading this post reminds me of my previous room mate!
He always kept chatting about this. I will forward
this post to him. Pretty sure he will have a good read.
Many thanks for sharing!
hi!,I really like your writing very so much! proportion we keep
in touch more about your post on AOL? I need a specialist
on this house to solve my problem. Maybe that is you!
Looking forward to look you.
Considering thɑt I started utiⅼizing DentiCore,
my oral ϲheck-uⲣs have actually been better. My teeth are stronger, and my
periodontals are healthier. The all-naturаl formula is mild yet efficient.
DentіCore is now a staple in my daily rօutіne.
My blog: Myrtis
Really informative blog article.Much thanks again. Fantastic.
I’ve tried numerous weight reduction supplements, but Nagano Lean Body Tonic sticks out. The mix of environment-friendly tea essence, ginger, and turmeric is powerful. It has actually helped me manage my cravings and increase my metabolic process. Plus, it’s simple to integrate right into my everyday routine. I’m thrilled with the outcomes!
Here is my blog post – https://opencbc.com/home.php?mod=space&uid=4488316
DentaVim is a fantastic item! My dental practitioner recommended it to help with my gum tissue problems, and I’ve seen amazing results. My gums are no longer puffy, and my breath remains fresh all day. It’s simple to take and fits perfectly into my daily routine. I could not be better with the results.
my page – https://martdaarad.com/profile/lynellsacco074
Very informative article post.Really thank you! Cool.
After trying various weight management supplements, ElectroSlim is the only one that has actually provided real results. This electrolyte drink not only aids with fat loss yet likewise maintains me hydrated and reduces my sugar food cravings. The all-natural, non-GMO, and gluten-free formula is a massive plus. I’ve shed 10 pounds in just a few weeks, and I feel much more energised than ever. ElectroSlim is a must-try for any individual serious concerning shedding weight.
Here is my homepage; https://Www.webwiki.fr/nativ.media:443/wiki/index.php?troutmother1985
Great article. Awesome.
ElectroSlim has surpassed my assumptions in every way. This electrolyte fat-burning beverage has not just helped me drop weight but likewise enhanced my overall wellness. The natural, non-GMO, and gluten-free active ingredients make it a risk-free and effective selection for fat burning. I’ve observed a substantial boost in my power degrees and a reduction in my sugar desires. ElectroSlim is the ideal service for anyone looking to shed pounds and remain healthy.
my web-site – https://Www.creativelive.com/student/raahauge-hwang?via=site-header_0
Siɡhtсare has actually really made a dіstinction in my life.
As a person who іnvests a great deal of time analysis and dealing ᴡith screens, I used to experience constant
eye fatigue and ⲣain. Because I started taking Sightсare, my
eyes really feel muсh more loosened up, and my vision is clearer.
The blend of vitamins and anti-oxidants in this supplement is fantastic.
I aⲣpreciate that it’s made from all-natural compߋnents, and I reɑlly feeⅼ ɡood
concerning taking іt each day. Highly suggest!
Feel free to surf to my blog post; Sightcare improve eyesight
Im grateful for the blog. Keep writing.
Its like you learn my thoughts! You seem to grasp a lot approximately this,
like you wrote the guide in it or something. I believe that you just could
do with a few percent to force the message home a little bit,
but instead of that, that is wonderful blog. A fantastic read.
I’ll certainly be back.
Nice blog right here! Additionally your website lots up fast! What web host are you the usage of? Can I get your affiliate hyperlink on your host? I desire my site loaded up as fast as yours lol.
Also visit my homepage http://www.yyml.online/bbs/home.php?mod=space&uid=1151825
I value the post.Much thanks again. Want more.
Ѕigһtcaгe has been a great addition to my wellness routine.
I staeted taking it to address the eyе strain I exprienced from long hours oof display time, and it has worked ԝonders.
My ѵision is sһarper, and I no ⅼonger really feel the ρain I used to.The natural components give me
assurance, understanding I’m supporting my eʏe health and wellness
without any kind of unsafe chemicals. Sightcare іs an еѕsentiаl for anybody wanting to keep healthy vision and mіnimize eye tiredness.
My webpage: Sightcare official website
ElectroSlim has been a game-changer for my weight loss journey. The combination of cravings reductions and increased energy degrees has made it less complicated to adhere to my diet plan and exercise regimen. I value that it’s created in an FDA-approved facility and has just natural, non-GMO, and gluten-free ingredients. If you’re having problem with weight management, provide ElectroSlim a shot– you won’t be dissatisfied!
Also visit my web blog – https://case.edu/cgi-bin/newsline.pl?URL=https://sady-spb.ru/user/nameclover16/
First of all I would like to say fantastic blog! I had a quick question that I’d like to ask if you do not mind. I was interested to find out how you center yourself and clear your head before writing. I’ve had a difficult time clearing my mind in getting my ideas out there. I truly do take pleasure in writing however it just seems like the first 10 to 15 minutes are generally lost just trying to figure out how to begin. Any recommendations or tips? Appreciate it!
Also visit my web page https://vuf.minagricultura.gov.co/Lists/Informacin%20Servicios%20Web/DispForm.aspx?ID=10358691
I seriously love your site.. Very nice colors & theme.
Did you make this amazing site yourself? Please reply
back as I’m attempting to create my own personal blog and would love
to know where you got this from or what the theme is called.
Cheers!
Hey just wanted to give you a quick heads up. The text in your content seem to be running off the screen in Internet explorer.
I’m not sure if this is a format issue or something to do with web browser compatibility but I figured I’d post to let you know.
The layout look great though! Hope you get the issue resolved soon. Thanks
You really make it appear so easy with your presentation but I to
find this matter to be really one thing which I think I would
by no means understand. It kind of feels too complex and extremely large for me.
I’m having a look forward to your subsequent publish, I will try to get the grasp of it!
Provadent is the ideal dental supplement I have actually attempted. My gum tissues utilized to hemorrhage frequently, yet since I started utilizing Provadent, they’ve become much healthier. The all-natural ingredients and probiotics job marvels. I can not envision my oral care regimen without it!
Feel free to visit my web blog: https://chiroqchi24.ru/user/zephyrmeal31/
I’ve been using VisiSoothe for some time now, and the enhancement in my vision is amazing. My eyes feel less strained, and I can see more clearly, also after lengthy hours of screen time. The all-natural formula is a large plus, and I enjoy that it’s designed to sustain general eye health and wellness. VisiSoothe is a superb item for anyone looking to improve their vision and eye health.
Take a look at my website :: http://Footballzaa.com/out.php?url=https://utahsyardsale.com/author/summerriver7/
Excellent items from you, man. I’ve bear in mind your stuff prior to and you’re simply extremely
fantastic. I really like what you have obtained right here, really like what you’re saying
and the way in which in which you are saying it. You are making it
enjoyable and you still care for to stay it smart.
I can’t wait to read far more from you. This is actually
a great website.
Hi, I do think this is a great blog. I stumbledupon it 😉 I may return once again since i have bookmarked it.
Money and freedom is the greatest way to change, may you be rich and continue to guide other people.
It’s amazing to pay a visit this site and reading the views of all friends
about this post, while I am also zealous of getting knowledge.
Hi there, i read your blog occasionally and i own a similar one and i was just curious if you get a lot of spam
responses? If so how do you prevent it, any plugin or anything you can recommend?
I get so much lately it’s driving me mad so any assistance is very much appreciated.
This text is worth everyone’s attention. When can I find out
more?
Really appreciate you sharing this blog article.Much thanks again. Will read on…
Nice post. I used to be checking constantly this blog and I am inspired!
Very helpful info specifically the closing part 🙂 I deal with such information much.
I was looking for this particular information for a long time.
Thanks and best of luck.
wow, awesome article.Thanks Again.
Hi there friends, how is the whole thing, and what you wish for to say concerning this article, in my view
its actually remarkable in favor of me.
As an adaptogen, Ashwagandha is made use of to ward off stress and anxiety exhaustion.
Check out my page :: http://123.249.110.128:5555/franziskaruthe
I’ve tried many oral products, but DentaVim is without a doubt the most effective. My teeth are whiter, my periodontals are healthier, and my breath is fresher. The all-natural formula is risk-free and effective, and I appreciate the high-quality production requirements. DentaVim is a must-have for any person serious concerning their oral wellness.
My web page; http://groszek.Katowice.pl/forum/profile.php?id=24651
ElectroSlim has actually exceeded my assumptions in every way. This electrolyte fat-burning drink has not only aided me slim down however likewise improved my total health and wellness. The natural, non-GMO, and gluten-free active ingredients make it a secure and efficient option for weight reduction. I have actually observed a substantial boost in my power degrees and a decrease in my sugar desires. ElectroSlim is the perfect remedy for any individual looking to drop extra pounds and stay healthy and balanced.
My page :: http://119.3.29.177:3000/manuelmacdonel
I’ve dealt with foul breath for years, and nothing appeared to work till I tried DentaVim. This supplement has actually totally changed my oral health regimen. My gums really feel more powerful, and my teeth are noticeably whiter. Plus, the truth that it’s made from natural ingredients is a big perk.
Here is my blog :: https://git.laser.Di.unimi.it/drusillafanny/natural-oral-hygiene-improvement1996/wiki/The-Relevance-of-Probiotics-in-Your-Dental-Wellness-Journey
I have actually observed a substantial decrease in plaque accumulation because I started making use of Provadent. My gum tissues are healthier, and my teeth feel stronger. The supplement is easy to take, and the outcomes are outstanding. Provadent is an excellent enhancement to my oral treatment routine.
Review my site; http://libochen.cn:13000/alda720159545/5090995/wiki/Exactly+How+to+Accomplish+Ideal+Dental+Care+with+Provadent+Products
I was cynical regarding vision supplements, yet VisiSoothe has surpassed my expectations. My vision has become more clear, and I feel much less stress after long days at job. The blend of natural components goes over, and I value the concentrate on eye health. VisiSoothe is a must-try for anyone experiencing vision concerns or desiring to maintain healthy eyes.
Feel free to surf to my web blog :: http://Recruitmentfromnepal.com/companies/visisoothe-web/
I love ProDentim! It has helped me accomplish a cleaner, fresher mouth. The probiotics in the formula sustain a balanced dental microbiome, reducing harmful bacteria. My breath is fresher, and my periodontals are healthier. ProDentim is a fantastic enhancement to my oral treatment regimen.
Visit my web site: http://dev.shopraves.com/minervarudolph
VisiSoothe has actually been a wonderful addition to my everyday routine. As a person who spends hours before a computer, my eyes were frequently exhausted and stretched. Given that starting VisiSoothe, I have actually observed a significant decrease in eye fatigue and improved quality. It’s a natural, reliable option for maintaining eye health. I can’t envision going back to life without it!
Feel free to surf to my homepage; https://Ima.it/en/
It is likewise found in the skin and may aid secure versus the sunlight’s destructive light.
Also visit my blog … http://Git.Nextopen.cn/avis1896373973/5382cellucare-glucose-support-supplement/-/issues/1
VisiSoothe has actually been a superb addition to my daily routine. As somebody who invests hours before a computer system, my eyes were constantly tired and stretched. Given that starting VisiSoothe, I’ve noticed a considerable reduction in eye exhaustion and enhanced clarity. It’s a natural, effective service for keeping eye wellness. I can not think of returning to life without it!
Feel free to surf to my web page; http://www.Thehispanicamerican.com/companies/visisoothe-web/
DentaVim is a lifesaver! My gums were constantly inflamed, and my breath was always bad. Considering that starting DentaVim, my periodontals have actually healed, and my breath is fresh all day long. The all-natural active ingredients offer me self-confidence that I’m taking treatment of my oral wellness in the very best method feasible.
Here is my website; https://git.Laser.di.unimi.it/hiram64q68859
Provadent is a fantastic item. My breath remains fresh all the time, and my teeth really feel stronger. The probiotics and all-natural ingredients are an excellent mix for keeping oral health. I highly advise Provadent to every person.
Here is my blog – http://47.76.210.186:3000/jasminegavin6
ElectroSlim has exceeded my expectations in every way. This electrolyte fat-burning beverage has not just helped me reduce weight but additionally boosted my total health and wellness. The natural, non-GMO, and gluten-free ingredients make it a risk-free and efficient choice for weight management. I’ve noticed a considerable boost in my energy levels and a decrease in my sugar food cravings. ElectroSlim is the best solution for any person seeking to shed pounds and stay healthy.
Feel free to visit my web blog https://Git.karma-Riuk.com/kerrifinniss0
Many cancer cells specialists suggest that you do not utilize vitamins or various other nutritional supplements.
my web blog – https://gitoa.ru/pearlinelutz51
Considering that beginning TonicGreens, I have actually observed a substantial renovation in my overall health. My food digestion is smoother, and I have extra endurance throughout the day. The fact that it’s gluten-free and non-GMO is a massive plus for me. TonicGreens is now a vital part of my day-to-day routine.
Here is my web-site :: https://gogs.kakaranet.com/deangelomaum1
I have actually been using Provadent for a month now, and the outcomes are wonderful. My breath is fresher, and I have less plaque build-up. The supplement is easy to integrate into my daily routine. Provadent is a game-changer for dental wellness!
Feel free to visit my web page – https://gitlab.ui.AC.Id/margaretwoodru
Today, we’ll discover 9 popularherbs and just how each can assist open up a brand-new chapter in health.
Also visit my web page: https://Sparktv.net/read-blog/14436_the-benefits-of-a-all-natural-supplement-like-cellucare-for-diabetics.html
Sightcare has been an amazing enhancement to my wellness regimen. I began taking it to attend to the eye stress I experienced from lengthy hours of screen time, and it has worked marvels. My vision is sharper, and I no more feel the pain I utilized to. The all-natural active ingredients provide me assurance, understanding I’m supporting my eye health and wellness without any type of hazardous chemicals. Sightcare is an essential for any person seeking to maintain healthy and balanced vision and decrease eye fatigue.
Also visit my web-site: http://git.decrunch.org/oliviaholyman1
Given that I started taking Keto Flow Gummies, my weight loss journey has actually been much smoother. These gummies are packed with BHB ketones that help my body melt fat for power. I’ve reduced weight, gained power, and decreased my food cravings. Keto Flow is an outstanding product for anyone on a keto diet. I can’t advise it enough!
Check out my page :: https://git.aerbim.com/patti798622494
What’s up to every , for the reason that I am really keen of reading this web site’s post to be updated daily.
It includes fastidious material.
Can I simply say what a comfort to find somebody that actually understands what they’re talking about on the net.
You certainly understand how to bring a problem to light and make it important.
More people have to check this out and understand this side of the
story. I was surprised that you are not more popular given that you surely have the gift.
You have made some decent points there. I looked on the internet for additional information about the issue and found
most individuals will go along with your views on this site.
May I just say what a relief to find someone that genuinely knows what they are talking about
online. You actually realize how to bring an issue to light and make
it important. A lot more people must read this and understand this side of the story.
I can’t believe you are not more popular given that you definitely possess the gift.
I love your blog.. very nice colors & theme. Did
you make this website yourself or did you hire someone to do it for you?
Plz respond as I’m looking to design my own blog
and would like to find out where u got this from. kudos
I used to be able to find good advice from your blog articles.
My family members every time say that I am wasting my time here at web, except
I know I am getting experience all the time by reading thes fastidious posts.
Im thankful for the blog article.Really looking forward to read more. Fantastic.
I truly appreciate this article.Much thanks again. Want more.
Thanks for finally writing about > 医薬におけるバリデーション
| お役立ち情報 < Liked it!
Say, you got a nice article.Much thanks again. Great.
I appreciate you sharing this article post.Really thank you! Much obliged.
Really informative blog.Thanks Again. Want more.
Have you ever considered about adding a little bit more than just your articles?
I mean, what you say is valuable and all. Nevertheless think of
if you added some great images or videos to give your posts more,
“pop”! Your content is excellent but with images
and videos, this site could undeniably be one of the best in its
niche. Awesome blog!
I have learn some good stuff here. Certainly price bookmarking for revisiting.
I surprise how a lot attempt you set to make this kind of magnificent informative site.
Hello, always i used to check weblog posts here in the early hours in the morning,
for the reason that i enjoy to find out more and more.
Pretty! This has been a really wonderful article.
Many thanks for providing these details.
Wonderful post! We are linking to this great content on our site.
Keep up the great writing.
I value the blog article. Fantastic.
Heya i am for the first time here. I came across this board and I in finding It
truly helpful & it helped me out much. I am hoping to present one
thing back and help others like you helped me.
Major thanks for the blog.Really looking forward to read more. Really Great.
Everyone loves what you guys are usually up too.
This type of clever work and exposure! Keep up the terrific works
guys I’ve you guys to my personal blogroll.
I truly appreciate this blog. Want more.
Great, thanks for sharing this post.Really thank you! Will read on…
This is one awesome blog article.Thanks Again. Really Great.
Hello! I’ve been reading your website for a while now
and finally got the bravery to go ahead and give you a shout out from Huffman Tx!
Just wanted to tell you keep up the fantastic work!
Your mode of explaining everything in this piece of writing is in fact fastidious, all can effortlessly know it, Thanks a lot.
Hi there, this weekend is pleasant designed for me, since this point in time i am reading this impressive educational piece of
writing here at my residence.
Major thanks for the post.Really thank you! Awesome.
Very good blog.Really thank you! Want more.
What’s up mates, how is all, and what you desire to say concerning this piece
of writing, in my view its truly remarkable designed for me.
I truly appreciate this article post.Really thank you! Keep writing.
Admiring the commitment you put into your website and detailed information you provide.
It’s good to come across a blog every once in a while that isn’t the same old rehashed information. Great
read! I’ve saved your site and I’m adding your RSS feeds to my Google account.
Very informative blog.Really thank you! Really Great.
This is one awesome blog post.Really thank you! Much obliged.
Thanks again for the blog post.Thanks Again. Great.
Just wish to say your article is as astonishing.
The clearness on your post is simply nice and i could suppose you’re a
professional on this subject. Fine along with
your permission let me to seize your RSS feed to stay updated with
coming near near post. Thank you 1,000,000 and please carry
on the gratifying work.
Hello There. I discovered your blog using msn. That is a very neatly written article.
I will be sure to bookmark it and come back to read extra of your useful information. Thanks for the post.
I’ll definitely comeback.
It’s enormous that you are getting thoughts from this post as well as from our
dialogue made here.
I do not even know how I ended up here, but I thought this post was great.
I do not know who you are but definitely you’re going to a famous blogger if you are not already 😉 Cheers!
Say, you got a nice post.Thanks Again. Will read on…
I really liked your blog post.Thanks Again. Keep writing.
Thanks to my father who told me regarding this website, this website
is actually amazing.
A round of applause for your blog article. Really Great.
Wow, great post. Much obliged.
I value the article post.Really thank you! Really Great.
I think this is a real great article post.Really looking forward to read more. Awesome.
wow, awesome post.Much thanks again.
What’s up, yeah this post is really good and I have learned lot of things from
it on the topic of blogging. thanks.
I truly appreciate this blog post.Thanks Again. Want more.
Im thankful for the post.Really thank you! Keep writing.
I really enjoy the blog post.Really thank you! Awesome.
Great blog.Really looking forward to read more. Will read on…
I am regular visitor, how are you everybody? This piece of writing
posted at this web page is genuinely good.
Really enjoyed this post.Really looking forward to read more. Cool.
After I initially commented I seem to have clicked the -Notify me when new comments are added- checkbox and from now on whenever a comment is
added I get 4 emails with the same comment.
Perhaps there is a means you can remove me from that service?
Kudos!
fantastic submit, very informative. I’m wondering why
the opposite experts of this sector don’t realize this.
You must continue your writing. I’m sure, you have a huge readers’ base already!
Really enjoyed this blog post.Really thank you! Keep writing.
I was wondering if you ever thought of changing the layout of your website?
Its very well written; I love what youve got to say.
But maybe you could a little more in the way of
content so people could connect with it better. Youve got an awful lot of text
for only having 1 or two pictures. Maybe you
could space it out better?
A big thank you for your blog. Awesome.
I truly appreciate this article post.Thanks Again. Keep writing.
Hello, yup this paragraph is actually nice and I have learned lot of
things from it about blogging. thanks.
I think this is a real great blog.Really looking forward to read more. Will read on…
Hmm it seems like your blog ate my first comment
(it was super long) so I guess I’ll just sum it up what I submitted and say, I’m thoroughly enjoying your blog.
I too am an aspiring blog writer but I’m still new to the whole thing.
Do you have any helpful hints for novice blog writers?
I’d genuinely appreciate it.
Everything is very open with a really clear explanation of the issues.
It was really informative. Your website is very useful. Thank you for sharing!
The other day, while I was at work, my cousin stole my iphone
and tested to see if it can survive a twenty five foot drop, just so she can be
a youtube sensation. My apple ipad is now destroyed and she has 83 views.
I know this is completely off topic but I had to share it with
someone!
Wow that was unusual. I just wrote an incredibly long comment but after
I clicked submit my comment didn’t appear. Grrrr…
well I’m not writing all that over again. Regardless, just
wanted to say superb blog!
Hi, I do think this is a great blog. I stumbledupon it 😉 I’m going to revisit once again since i have book-marked
it. Money and freedom is the best way to change, may you be rich and continue to help others.
I am not sure where you’re getting your info, but good topic.
I needs to spend some time learning more or understanding more.
Thanks for magnificent info I was looking for this info for my mission.
Fantastic beat ! I wish to apprentice at the same time as you amend your web
site, how can i subscribe for a blog site? The account
aided me a appropriate deal. I were tiny bit acquainted of this your broadcast provided vivid transparent concept
Thanks-a-mundo for the article post.Really looking forward to read more. Fantastic.
Pretty! This was an extremely wonderful article. Thanks for supplying this
info.
I always used to read piece of writing in news
papers but now as I am a user of net therefore from now I am using
net for posts, thanks to web.
Hi everyone, it’s my first pay a quick visit at this
web site, and piece of writing is truly fruitful in support of me, keep up posting such articles or reviews.
I’m extremely impressed with your writing skills as well as with the layout
on your blog. Is this a paid theme or did you modify it yourself?
Anyway keep up the nice quality writing, it’s rare to see a nice blog like this one these days.
If some one desires to be updated with hottest
technologies after that he must be go to see this site and be up to
date every day.
Hi my friend! I want to say that this post is awesome, great written and come with
approximately all significant infos. I would like to see extra posts like
this .
What a material of un-ambiguity and preserveness of precious
know-how concerning unexpected feelings.
Hi there just wanted to give you a quick heads up.
The text in your post seem to be running off the screen in Firefox.
I’m not sure if this is a formatting issue or something to do with browser compatibility
but I thought I’d post to let you know. The layout look great
though! Hope you get the problem resolved soon. Many thanks
Link exchange is nothing else except it is only placing the other person’s
website link on your page at suitable place and other person will also do same in favor of you.
Oh my goodness! Impressive article dude! Many thanks, However I am encountering difficulties with your RSS.
I don’t know the reason why I can’t join it. Is there anyone else getting identical RSS problems?
Anyone that knows the answer can you kindly respond? Thanx!!
Heya i am for the first time here. I found this board and I find It truly useful & it helped
me out much. I hope to give something back and help
others like you helped me.
hello there and thank you for your information – I’ve
definitely picked up anything new from right here. I did
however expertise several technical issues using this web site, since
I experienced to reload the web site lots of times previous to I could get it to load properly.
I had been wondering if your hosting is OK? Not that I am complaining,
but sluggish loading instances times will often affect
your placement in google and could damage your quality score if advertising and marketing with Adwords.
Anyway I’m adding this RSS to my email and can look out
for a lot more of your respective exciting content.
Make sure you update this again very soon.
Looking forward to reading more. Great article post.Thanks Again. Cool.
Thank you for your article.Thanks Again.
Really appreciate you sharing this post.Really looking forward to read more.
Really informative article. Want more.
It is perfect time to make a few plans for the future and it is time to be happy.
I’ve read this put up and if I could I wish to suggest you some attention-grabbing things
or advice. Maybe you can write next articles regarding this article.
I wish to learn even more issues approximately it!
Im thankful for the blog post. Awesome.
Looking forward to reading more. Great blog article.Really thank you!
I value the blog post.Really looking forward to read more. Awesome.
Greetings! I’ve been reading your blog for some time now and finally got the bravery to go ahead and give you a shout out from Austin Texas!
Just wanted to mention keep up the excellent work!
We are looking for some people that might be interested in from working their home on a full-time basis. If you want to earn $500 a day, and you don’t mind writing some short opinions up, this might be perfect opportunity for you!
We are searching for some people that might be interested in from working their home on a full-time basis. If you want to earn $200 a day, and you don’t mind creating some short opinions up, this is the perfect opportunity for you!
At this moment I am going to do my breakfast, afterward having my breakfast coming again to read additional news.
Great, thanks for sharing this blog.Really thank you! Keep writing.
Hello, i feel that i noticed you visited my site so i came to return the desire?.I am attempting to to find issues to improve my
web site!I suppose its adequate to make use of some
of your ideas!!
When someone writes an paragraph he/she maintains the idea of a user in his/her mind that how a
user can know it. Thus that’s why this piece of writing is
outstdanding. Thanks!
Awesome blog article.Really thank you! Keep writing.
I cannot thank you enough for the blog article.Really looking forward to read more. Will read on…
This post is priceless. How can I find out more?
Great, thanks for sharing this blog.Thanks Again.
I’m not sure exactly why but this site is loading extremely slow for
me. Is anyone else having this problem or is it a problem on my end?
I’ll check back later on and see if the problem still exists.
I think this is a real great post.Much thanks again. Great.
Im obliged for the article post.Really looking forward to read more. Keep writing.
Great article.Really thank you! Really Great.
Im obliged for the article post.Thanks Again. Really Great.
Hello there! I just would like to offer you a big thumbs up for the great information you have right here on this post.
I will be returning to your blog for more soon.
I really enjoy the blog.Really looking forward to read more. Cool.
Major thankies for the blog.Really looking forward to read more. Cool.
This post will assist the internet users for creating new website or even a weblog from start to end.
Im obliged for the blog.Really looking forward to read more. Fantastic.
This is a topic which is near to my heart… Take care!
Where are your contact details though?
Im obliged for the blog.Thanks Again. Awesome.
Valuable info. Fortunate me I discovered your web site unintentionally, and I’m stunned
why this coincidence didn’t took place earlier! I bookmarked
it.
Hi there, this weekend is pleasant in favor of me, for the
reason that this point in time i am reading this great informative
piece of writing here at my house.
I’m gone to inform my little brother, that he should also go
to see this website on regular basis to take updated from most up-to-date reports.
Hey, I think your blog might be having browser compatibility issues.
When I look at your website in Ie, it looks fine but when opening in Internet Explorer,
it has some overlapping. I just wanted to give you a quick heads up!
Other then that, very good blog!
Hi, i think that i saw you visited my website so i got here to go back the prefer?.I am trying to
in finding issues to enhance my web site!I guess its adequate
to use some of your concepts!!
It’s an remarkable post designed for all the internet viewers; they will obtain advantage from it I am sure.
Normally I do not learn post on blogs, however
I would like to say that this write-up very compelled me to take a look
at and do so! Your writing style has been amazed me.
Thanks, quite nice article.
I got this site from my friend who shared with me about
this website and at the moment this time I am visiting this
web page and reading very informative posts at this time.
Great post.Really thank you! Keep writing.
I am really enjoying the theme/design of your web site.
Do you ever run into any internet browser compatibility problems?
A few of my blog audience have complained about my
website not working correctly in Explorer but looks great in Firefox.
Do you have any suggestions to help fix this issue?
Awesome post.Really looking forward to read more. Cool.
I value the blog article.Thanks Again. Awesome.
I read this paragraph completely on the topic of the resemblance of newest and
previous technologies, it’s remarkable article.
Thanks so much for the blog article.Really looking forward to read more. Keep writing.
Good post. I learn something totally new and challenging on sites I stumbleupon on a daily basis.
It will always be exciting to read through articles from other
writers and practice something from other websites.
Very neat article post.Thanks Again. Much obliged.
Really appreciate you sharing this article.Really thank you! Awesome.
Major thanks for the post.Thanks Again. Really Great.
Im obliged for the blog article.Thanks Again. Will read on…
Hello! I understand this is somewhat off-topic but I had to ask.
Does building a well-established website such as yours take a large amount of work?
I’m completely new to blogging but I do write in my diary everyday.
I’d like to start a blog so I will be able to share my personal experience and thoughts online.
Please let me know if you have any kind of ideas or
tips for new aspiring bloggers. Appreciate it!
Hey very interesting blog!
It’s actually a cool and useful piece of information. I am happy that you simply shared this useful info with us.
Please keep us up to date like this. Thanks for sharing.
This is a very good tip especially to those new to the blogosphere.
Short but very accurate info… Many thanks for sharing this one.
A must read article!
Muchos Gracias for your article post.Really thank you! Keep writing.
Very neat blog article. Cool.
I’m truly enjoying the design and layout of your blog.
It’s a very easy on the eyes which makes it much more pleasant for me
to come here and visit more often. Did you hire out a developer to create your
theme? Exceptional work!
I really liked your blog article.Really thank you! Much obliged.
Hey, thanks for the article.Really thank you! Great.
Hey, thanks for the article post.Really thank you! Want more.
Thanks for the article post.Really thank you! Cool.
Article writing is also a fun, if you be acquainted with afterward
you can write otherwise it is complex to write.
Im thankful for the article post.Really thank you! Awesome.
A big thank you for your blog.Thanks Again.
Thanks again for the blog post.Thanks Again.
Hello! I realize this is kind of off-topic however I needed to ask.
Does running a well-established website such as yours take
a large amount of work? I’m brand new to operating a blog but I do write in my diary every day.
I’d like to start a blog so I will be able to share my experience and
feelings online. Please let me know if you have any
ideas or tips for brand new aspiring bloggers. Appreciate it!
I’m not positive where you are getting your information,
however great topic. I needs to spend a while finding out much more or figuring
out more. Thank you for wonderful info I used to be searching for this info for my mission.
Hey, I think your site might be having browser compatibility issues.
When I look at your blog in Safari, it looks fine but when opening in Internet Explorer, it has some overlapping.
I just wanted to give you a quick heads up! Other then that, very good blog!
I think this is a real great article post.Thanks Again. Cool.
I cannot thank you enough for the article.Really thank you! Cool.
Thank you for your blog.Really looking forward to read more. Really Cool.
wow, awesome blog.Much thanks again. Will read on…
I appreciate you sharing this blog article.Much thanks again. Keep writing.
I’ve learn several good stuff here. Certainly price bookmarking for revisiting.
I surprise how much effort you put to create the sort of excellent
informative site.
Really informative article post.Thanks Again. Keep writing.
I really like and appreciate your blog.Really thank you! Will read on…
Great article, just what I was looking for.
Sweet blog! I found it while surfing around on Yahoo News.
Do you have any tips on how to get listed in Yahoo News?
I’ve been trying for a while but I never seem to get there!
Thanks
I value the post.Really thank you! Really Great.
Very neat blog.Really thank you! Fantastic.
Thanks for sharing, this is a fantastic article post.Much thanks again. Want more.
Im grateful for the post.Thanks Again. Great.
Really informative blog.Really thank you! Really Great.
I truly appreciate this blog.Really looking forward to read more. Great.
I visited multiple websites however the audio feature for
audio songs present at this web site is genuinely wonderful.
A big thank you for your blog.Much thanks again. Really Cool.
Pretty nice post. I just stumbled upon your weblog and wanted to
say that I’ve truly enjoyed surfing around your blog posts.
After all I will be subscribing to your feed and I hope you write again soon!
I value the post. Great.
Thanks for sharing your info. I really appreciate your efforts and I am waiting for your further post thank you once again.
Really appreciate you sharing this blog post.Thanks Again. Great.
DentaVim has made an obvious distinction in my dental hygiene. My breath is fresher, and my periodontals are no more bleeding when I comb. The blend of natural components works wonders, and I value the high production criteria. This item deserves every cent!
Stop by my webpage :: http://www.hjvalve.co.kr/bbs/board.php?bo_table=free&wr_id=524759
Wonderful, what a blog it is! This website presents helpful information to
us, keep it up.
Hi everyone, it’s my first visit at this site, and post is in fact fruitful in support of me,
keep up posting such articles.
ElectroSlim has surpassed my assumptions in every way. This electrolyte fat-burning drink has not just helped me reduce weight however likewise boosted my general wellness. The natural, non-GMO, and gluten-free components make it a risk-free and reliable choice for weight reduction. I’ve observed a considerable increase in my energy degrees and a reduction in my sugar food cravings. ElectroSlim is the best remedy for any person wanting to shed pounds and stay healthy.
Here is my blog; https://Asesordocente.com/question/healthy-way-of-life-hacks-for-a-fitter-happier-you/
TonicGreens has been a game-changer for me. I enjoy that it’s all-natural and made in the USA. Considering that I began taking it, my digestion has boosted, and I really feel a lot more invigorated. It’s currently a staple in my daily wellness routine. Wonderful product!
Feel free to visit my web-site https://Skillsinternational.co.in/employer/tonicgreens-ltd/
Muchos Gracias for your post.Much thanks again. Really Great.
VisiSoothe has been a great addition to my day-to-day regimen. As somebody who spends hours in front of a computer, my eyes were constantly tired and strained. Considering that starting VisiSoothe, I have actually discovered a considerable decrease in eye tiredness and enhanced clarity. It’s an all-natural, effective service for keeping eye health. I can’t visualize returning to life without it!
my website … http://newborhooddates.com/@adelapresley76
I value the article post.Really looking forward to read more. Fantastic.
Why people still use to read news papers when in this technological world all is accessible
on net?
Great, thanks for sharing this blog article.Much thanks again. Much obliged.
Really enjoyed this article post.Really looking forward to read more. Will read on…
Major thankies for the blog.Really thank you! Keep writing.
Howdy very nice blog!! Man .. Beautiful .. Wonderful ..
I’ll bookmark your website and take the feeds also?
I am glad to search out so many useful information here in the publish,
we need work out more techniques on this regard, thanks for sharing.
. . . . .
Really appreciate you sharing this article post.Really looking forward to read more. Awesome.
Awesome post.Really thank you! Will read on…
I loved your article. Awesome.
wow, awesome article.Really thank you! Keep writing.
Great post.Really thank you! Really Cool.
Hey There. I found your weblog the use of msn. This is
an extremely smartly written article. I’ll be sure to bookmark it and return to read more of your helpful info.
Thank you for the post. I’ll definitely return.
Amazing blog! Do you have any recommendations for aspiring writers?
I’m hoping to start my own website soon but I’m a little lost on everything.
Would you suggest starting with a free platform like WordPress or go for a paid option?
There are so many choices out there that I’m totally confused ..
Any suggestions? Thanks a lot!
I am so grateful for your blog article.Much thanks again. Really Great.
Thank you ever so for you blog article.Really thank you! Great.
Greetings! I know this is kinda off topic nevertheless I’d
figured I’d ask. Would you be interested in exchanging links or maybe
guest writing a blog article or vice-versa?
My site discusses a lot of the same subjects as yours and I believe we could greatly benefit from each other.
If you happen to be interested feel free to shoot me an email.
I look forward to hearing from you! Superb blog by the way!
Thanks so much for the post.Really thank you! Want more.
Great, thanks for sharing this article post.Really thank you! Want more.
great points altogether, you simply received a brand new reader.
What may you suggest about your put up that you just made
some days ago? Any sure?
My partner and I stumbled over here by a different page and thought I might as well check things out.
I like what I see so now i am following you. Look forward to exploring your web page for a second time.
A round of applause for your blog.Really thank you! Cool.
Thanks again for the article.Really looking forward to read more. Great.
Say, you got a nice article post. Keep writing.
Hi would you mind letting me know which web host you’re working
with? I’ve loaded your blog in 3 different internet browsers
and I must say this blog loads a lot faster then most.
Can you suggest a good hosting provider at a fair price?
Thanks, I appreciate it!
I am so grateful for your blog article.Thanks Again. Will read on…
Major thanks for the blog article.Really looking forward to read more. Keep writing.
I think this is one of the most significant info for me.
And i am glad reading your article. But wanna remark on some general things, The web site
style is perfect, the articles is really great : D. Good job, cheers
Hey There. I found your blog using msn. This is a very well
written article. I’ll be sure to bookmark it and return to read more of your
useful information. Thanks for the post. I will definitely comeback.
Thanks a lot for the blog article.Much thanks again.
There is certainly a great deal to find out about this subject.
I like all the points you have made.
Thanks for the article.Thanks Again. Great.
Awesome post. Will read on…
I really like and appreciate your blog article.Much thanks again. Really Great.
Thank you ever so for you article post.Really thank you! Really Great.
Say, you got a nice article post.Much thanks again. Want more.
Hello there! Would you mind if I share your blog with my twitter group?
There’s a lot of people that I think would really appreciate your content.
Please let me know. Many thanks
I don’t even understand how I ended up here, however I assumed
this submit used to be great. I don’t realize who you might be but
definitely you’re going to a famous blogger if you are not already.
Cheers!
Thanks for the article.Really thank you! Great.
Major thanks for the blog post.Really thank you! Will read on…
Major thankies for the post.Really thank you! Really Great.
Awesome blog.Thanks Again. Much obliged.
Excellent article. I am facing some of these issues as well..
I really enjoy the article.Much thanks again. Great.
A big thank you for your blog post.Thanks Again. Much obliged.
I loved your post. Keep writing.
I love your blog.. very nice colors & theme. Did you design this website yourself or
did you hire someone to do it for you? Plz respond as I’m looking to create my own blog and would like to know where
u got this from. cheers
Muchos Gracias for your article.Really thank you! Great.
I’m truly enjoying the design and layout of your website.
It’s a very easy on the eyes which makes it much more enjoyable for
me to come here and visit more often. Did you hire out a developer to create your theme?
Exceptional work!
This site was… how do you say it? Relevant!! Finally I’ve found something that helped me.
Thanks!
Very good blog post.Much thanks again.
Hey, thanks for the blog. Really Cool.
I really liked your blog post. Cool.
I cannot thank you enough for the post.Really thank you! Much obliged.
Im obliged for the article.Much thanks again. Great.
each time i used to read smaller content that also clear their motive, and that is also happening with this article which I am reading here.
Very quickly this web page will be famous among all blogging people,
due to it’s good content
I appreciate you sharing this article post. Cool.
Great, thanks for sharing this blog article.Thanks Again. Fantastic.
Oh my goodness! Impressive article dude! Many thanks, However
I am going through issues with your RSS. I don’t understand why I can’t subscribe to it.
Is there anyone else getting similar RSS problems?
Anybody who knows the solution will you kindly respond?
Thanks!!
Thank you ever so for you blog article.Really thank you! Great.
I really enjoy the blog article.Thanks Again.
It’s hard to find experienced people for this
topic, but you sound like you know what you’re talking about!
Thanks
This is one awesome article.Thanks Again. Want more.
I truly appreciate this article.Thanks Again. Great.
Enjoyed every bit of your blog article.Thanks Again. Cool.
I really enjoy the article.Really thank you! Keep writing.
Very informative blog.Really thank you!
Wow that was odd. I just wrote an very long comment but after I clicked submit my comment
didn’t appear. Grrrr… well I’m not writing all that over again. Regardless,
just wanted to say great blog!
What’s up every one, here every person is sharing such
knowledge, so it’s fastidious to read this website, and I used to pay a visit this
blog daily.
Hey great blog! Does running a blog such as this take a lot
of work? I have virtually no understanding of coding but I
had been hoping to start my own blog soon. Anyhow, if you have any suggestions or tips for new blog owners please share.
I know this is off subject however I simply had to ask.
Appreciate it!
What’s Happening i’m new to this, I stumbled upon this I have found It
positively helpful and it has helped me out loads.
I hope to contribute & assist different customers like its helped me.
Great job.
Have you ever thought about adding a little bit more than just your articles?
I mean, what you say is valuable and everything.
Nevertheless think about if you added some great visuals or videos
to give your posts more, “pop”! Your content is excellent but with pics and
videos, this blog could certainly be one of the most beneficial
in its field. Good blog!
I have read so many posts regarding the blogger lovers but this paragraph
is genuinely a nice paragraph, keep it up.
Hi there, every time i used to check weblog posts here in the
early hours in the break of day, for the reason that i love to gain knowledge of more
and more.
Im obliged for the blog post.Thanks Again. Really Great.
Im grateful for the article. Cool.
Howdy! I know this is somewhat off topic but
I was wondering which blog platform are you using for this site?
I’m getting tired of WordPress because I’ve had problems with hackers and I’m looking at options for
another platform. I would be fantastic if you could point me in the direction of a
good platform.
Very informative article.Really looking forward to read more. Keep writing.
Say, you got a nice blog.Much thanks again. Awesome.
I appreciate you sharing this blog post.Much thanks again. Cool.
Hi! This post couldn’t be written any better! Reading through this post reminds me of my previous room mate!
He always kept chatting about this. I will forward
this write-up to him. Fairly certain he will have a good read.
Thanks for sharing!
Hi! I’m at work surfing around your blog from my new apple iphone!
Just wanted to say I love reading your blog and look forward to all your posts!
Carry on the great work!
hey there and thank you for your info – I have certainly picked up something new
from right here. I did however expertise several technical
issues using this site, as I experienced to reload the web site lots of times previous to
I could get it to load properly. I had been wondering if your hosting is OK?
Not that I’m complaining, but sluggish loading
instances times will very frequently affect your placement in google and can damage your quality score if advertising and marketing with Adwords.
Well I’m adding this RSS to my e-mail and could look out for much more of your respective intriguing
content. Ensure that you update this again soon.
I delight in, lead to I found just what I was having a look
for. You’ve ended my 4 day lengthy hunt! God Bless you man. Have a
great day. Bye
Hello, Neat post. There’s a problem with your web site in internet explorer,
would check this? IE nonetheless is the marketplace leader
and a huge component of other folks will miss your great writing due to this
problem.
I don’t even know how I ended up here, but I thought this post was great.
I do not know who you are but certainly you’re going to
a famous blogger if you are not already 😉 Cheers!
Wonderful blog! I found it while browsing on Yahoo News. Do
you have any tips on how to get listed in Yahoo News?
I’ve been trying for a while but I never seem to get there!
Thanks
Cool blog! Is your theme custom made or did you download it from somewhere?
A theme like yours with a few simple tweeks would really
make my blog stand out. Please let me know where you got your design. Appreciate it
Great blog! Is your theme custom made or did you download it from somewhere?
A theme like yours with a few simple tweeks would really make my blog stand out.
Please let me know where you got your theme. Many thanks
It’s amazing designed for me to have a website, which is good for my
experience. thanks admin
Thanks for some other informative blog. Where else may I am getting that kind of info written in such an ideal method?
I have a venture that I am simply now working on, and I’ve been at
the glance out for such information.
Hmm it appears like your website ate my first comment (it was super long) so I guess I’ll just sum it up what I submitted and say, I’m thoroughly enjoying your blog.
I too am an aspiring blog writer but I’m still
new to the whole thing. Do you have any helpful hints for novice blog writers?
I’d genuinely appreciate it.
Ridiculous quest there. What occurred after? Take care!
Hello there! I simply would like to offer you a huge thumbs up for the great info you have here on this post.
I will be returning to your web site for more soon.
Hi there everyone, it’s my first pay a visit at this site, and piece
of writing is really fruitful for me, keep up posting such posts.
whoah this blog is fantastic i like studying your posts.
Stay up the good work! You realize, a lot of individuals are looking round for this information, you can help them greatly.
Thank you for any other informative site. The place else could I
am getting that type of info written in such a perfect
manner? I have a challenge that I’m just now working on, and
I’ve been at the look out for such info.
Hey there! Do you use Twitter? I’d like to follow you
if that would be ok. I’m definitely enjoying your blog and look forward
to new posts.
This site was… how do I say it? Relevant!! Finally I have found something which helped me.
Appreciate it!
It’s an remarkable paragraph in support of all the web users; they
will obtain advantage from it I am sure.
Wow, incredible blog layout! How long have you been blogging for?
you made blogging look easy. The overall look of your website is magnificent, let alone the
content!
I’m really enjoying the design and layout of
your website. It’s a very easy on the eyes which makes it much more pleasant for me to come here and
visit more often. Did you hire out a designer to create your theme?
Superb work!
Valuable info. Lucky me I found your site unintentionally, and I am shocked why this twist of fate didn’t came about in advance!
I bookmarked it.
I was curious if you ever thought of changing the page
layout of your site? Its very well written; I love what youve got to
say. But maybe you could a little more in the way of content so
people could connect with it better. Youve got an awful lot of text for only having 1 or 2 images.
Maybe you could space it out better?
I have been surfing online more than three hours
today, yet I never found any interesting article like yours.
It’s pretty worth enough for me. In my opinion, if all
webmasters and bloggers made good content as you did, the internet will be a lot more useful than ever before.
Please let me know if you’re looking for a writer for your blog.
You have some really good posts and I believe I would be a good asset.
If you ever want to take some of the load off,
I’d love to write some articles for your blog in exchange for a link back to mine.
Please shoot me an e-mail if interested. Kudos!
Wow! In the end I got a website from where I can truly get helpful facts concerning my study and
knowledge.
Undeniably believe that which you stated. Your favorite reason seemed to be on the internet
the simplest thing to be aware of. I say to you, I definitely get irked while people think about worries that they just do
not know about. You managed to hit the nail upon the top as well as defined out
the whole thing without having side-effects , people could take a signal.
Will probably be back to get more. Thanks
What’s up, the whole thing is going nicely here
and ofcourse every one is sharing information, that’s really excellent, keep up
writing.
That is very interesting, You are a very professional
blogger. I’ve joined your rss feed and look ahead to in quest of extra
of your fantastic post. Additionally, I’ve shared your site in my social networks
Simply want to say your article is as astounding. The clarity
in your post is simply spectacular and i could assume you’re an expert on this subject.
Fine with your permission let me to grab your feed to keep up to date with forthcoming post.
Thanks a million and please continue the rewarding work.
Greetings! Very helpful advice in this particular post!
It’s the little changes that produce the most significant
changes. Thanks for sharing!
Do you have any video of that? I’d care to find out more details.
I absolutely love your website.. Pleasant colors
& theme. Did you develop this web site yourself?
Please reply back as I’m planning to create my own site
and want to learn where you got this from or exactly what the theme is named.
Many thanks!
Aw, this was a very nice post. Taking the time and actual effort to create a
great article… but what can I say… I hesitate a lot
and never seem to get nearly anything done.
Hey there just wanted to give you a quick heads up.
The text in your content seem to be running off the screen in Opera.
I’m not sure if this is a format issue or something to do with web browser compatibility but I figured I’d
post to let you know. The design look great though!
Hope you get the problem resolved soon. Thanks
With havin so much written content do you ever run into any problems of plagorism or
copyright violation? My blog has a lot of exclusive content I’ve either written myself or outsourced but it appears a lot of it
is popping it up all over the web without my permission. Do you
know any methods to help stop content from being stolen? I’d
truly appreciate it.
I love what you guys are usually up too. This sort of clever work and coverage!
Keep up the very good works guys I’ve incorporated you guys
to my blogroll.
Way cool! Some extremely valid points! I appreciate you penning this article and the rest of the site is very good.
What’s up to all, because I am really eager of reading this weblog’s
post to be updated daily. It carries pleasant data.
This is very interesting, You are a very skilled blogger.
I’ve joined your rss feed and look forward to seeking more of
your great post. Also, I’ve shared your website in my social networks!
Hi, I think your website might be having browser compatibility issues.
When I look at your blog in Ie, it looks fine but when opening in Internet Explorer, it has some overlapping.
I just wanted to give you a quick heads up! Other then that, wonderful blog!
I’m not sure exactly why but this site is loading incredibly slow
for me. Is anyone else having this issue or is it a problem on my end?
I’ll check back later and see if the problem still exists.
Hi I am so thrilled I found your site, I really found you by mistake,
while I was browsing on Askjeeve for something else, Anyways I am here now and would just
like to say thanks a lot for a incredible post and a all
round entertaining blog (I also love the theme/design), I don’t have time to look over
it all at the minute but I have bookmarked it and also added your RSS feeds, so when I have time I will
be back to read more, Please do keep up the superb jo.
Thanks for finally talking about > 医薬におけるバリデーション | お役立ち情報 < Liked it!
Please let me know if you’re looking for a article writer
for your weblog. You have some really great articles and I feel I would be a good asset.
If you ever want to take some of the load off, I’d absolutely love to write some material for your blog in exchange for a
link back to mine. Please send me an email if interested.
Kudos!
I am truly delighted to read this webpage posts which includes plenty of helpful facts,
thanks for providing these kinds of information.
I was recommended this web site by my cousin. I’m not
sure whether this post is written by him as nobody else know
such detailed about my problem. You are incredible! Thanks!
Excellent post. I was checking continuously this blog and
I am impressed! Extremely useful info specifically the last part 🙂 I care for such
information much. I was looking for this particular info for a very long time.
Thank you and good luck.
This is really interesting, You are a very skilled blogger.
I’ve joined your feed and look forward to seeking more of
your fantastic post. Also, I’ve shared your
website in my social networks!
It is not my first time to go to see this website, i am visiting this website dailly and get good
information from here everyday.
I have been exploring for a bit for any high-quality articles or blog posts in this
kind of space . Exploring in Yahoo I at last stumbled upon this web site.
Reading this info So i’m glad to convey that I’ve a very good uncanny feeling I came upon exactly what I needed.
I such a lot for sure will make sure to do not disregard this site and
give it a look regularly.
If you are going for best contents like me, just pay a quick visit this website every day because it offers quality
contents, thanks
Hi just wanted to give you a brief heads up and let
you know a few of the pictures aren’t loading correctly.
I’m not sure why but I think its a linking issue. I’ve tried it in two
different internet browsers and both show the same
results.
This post gives clear idea in favor of the new viewers of blogging, that in fact how to do blogging and site-building.
I really like your blog.. very nice colors & theme. Did you make this website yourself or did you hire
someone to do it for you? Plz reply as I’m looking to create
my own blog and would like to know where u got this from.
cheers
I think that what you wrote was actually very reasonable.
However, consider this, suppose you added a little information? I mean,
I don’t wish to tell you how to run your website,
however suppose you added a title that grabbed folk’s attention? I mean 医薬におけるバリデーション | お役立ち情報
is kinda boring. You should look at Yahoo’s front page and see how they create news
titles to get viewers interested. You might add a
video or a pic or two to grab people interested about everything’ve got to say.
In my opinion, it would bring your posts a little livelier.
Remarkable issues here. I’m very satisfied to look your post.
Thank you a lot and I am having a look ahead to touch you.
Will you please drop me a e-mail?
Hi, this weekend is nice designed for me, because this moment i am reading
this enormous informative post here at my house.
Thanks for sharing your thoughts on iconet computer. Regards
I am curious to find out what blog system you have been using?
I’m experiencing some small security issues with my latest blog
and I’d like to find something more secure. Do you have any suggestions?
I do not know if it’s just me or if perhaps everybody else experiencing problems with your blog.
It looks like some of the text within your content are running off the screen. Can somebody else please provide feedback
and let me know if this is happening to them too? This may be a issue with my
web browser because I’ve had this happen before. Many thanks
Every weekend i used to pay a quick visit this web page, as i wish for enjoyment, as this this site conations really fastidious funny information too.
Thanks for information Kehidupan Kampus
Attractive section of content. I just stumbled upon your web site and in accession capital to assert that
I acquire in fact enjoyed account your blog posts.
Anyway I will be subscribing to your feeds and even I achievement you
access consistently fast.
These are truly great ideas in about blogging. You have touched some
pleasant points here. Any way keep up wrinting.
Whats up very nice blog!! Guy .. Beautiful .. Wonderful ..
I’ll bookmark your blog and take the feeds also?
I am glad to seek out so many useful information right here
within the post, we need develop extra strategies
in this regard, thanks for sharing. . . . . .
I believe what you posted was very logical.
But, think about this, suppose you added a little content?
I ain’t saying your content is not solid, however what if you added a post title to possibly grab a person’s attention? I mean 医薬におけるバリデーション | お役立ち情報 is a little plain. You could glance at Yahoo’s home page and note how they write
news titles to grab viewers to click. You might add a video or a pic or two to
get readers excited about everything’ve written. Just my opinion, it would bring your posts a little bit more interesting.
I think the admin of this web site is actually working
hard for his web site, because here every material is quality based information.
Write more, thats all I have to say. Literally,
it seems as though you relied on the video to make your point.
You definitely know what youre talking about, why waste your
intelligence on just posting videos to your weblog when you could be giving us something enlightening to read?
Pretty section of content. I just stumbled upon your weblog and in accession capital to assert that I get actually enjoyed account
your blog posts. Anyway I will be subscribing to your augment and even I achievement you access consistently quickly.
Greetings! Very helpful advice in this particular article!
It is the little changes that produce the greatest changes.
Thanks for sharing!
Hi there, I read your blogs on a regular basis. Your story-telling
style is awesome, keep doing what you’re doing!
I am genuinely pleased to glance at this blog posts which includes lots of helpful data, thanks for
providing such information.
Wonderful web site. Plenty of useful information here. I am sending it to
several buddies ans additionally sharing in delicious.
And of course, thanks in your sweat!
Hi there colleagues, pleasant paragraph and nice arguments commented at this place, I am
actually enjoying by these.
I’m amazed, I must say. Seldom do I come across a blog that’s both educative and entertaining, and let me tell you, you’ve hit the nail on the head.
The problem is something which too few men and women are speaking
intelligently about. I’m very happy I stumbled across this during my search for something concerning this.
Thanks for sharing your thoughts. I truly appreciate your efforts and I will
be waiting for your further post thank you once again.
We are one of the official gacor slot platforms that you can visit right now.
Howdy! Someone in my Facebook group shared
this site with us so I came to give it a look.
I’m definitely loving the information. I’m book-marking and will
be tweeting this to my followers! Fantastic blog and great design.
Unquestionably believe that which you stated.
Your favorite justification seemed to be on the internet the simplest thing to be aware
of. I say to you, I certainly get irked while people think about worries that they just don’t
know about. You managed to hit the nail upon the top and
also defined out the whole thing without having side-effects ,
people can take a signal. Will probably be back to get more.
Thanks
If some one needs expert view about blogging and site-building after that i recommend him/her to visit this website, Keep up the pleasant job.
Heya i am for the first time here. I came across this board and I find It really helpful & it helped me out
much. I am hoping to provide something again and aid others such as you aided me.
Tamamen dolandırıcılık yapıyor, insanlar buraya gelmesin, ben bu sitede 100k$ kaybettim: Japon pornoları
Hey there! I’ve been reading your site for a long time now and finally got the bravery to go ahead and give you a shout out from Humble Texas!
Just wanted to tell you keep up the good work!
You ought to be a part of a contest for one of the finest blogs
on the net. I most certainly will recommend this blog!
You’ll Be Unable To Guess Sofa Savings’s Benefits Sofa savings
Greetings from Los angeles! I’m bored at work so I decided to check out your site
on my iphone during lunch break. I enjoy the info you present here and can’t wait
to take a look when I get home. I’m surprised at how fast
your blog loaded on my cell phone .. I’m not even using WIFI,
just 3G .. Anyways, awesome blog!
Guide To Best Robot Vacuum Uk: The Intermediate Guide To Best Robot Vacuum Uk best robot vacuum uk
5 Killer Quora Answers To Adult Mens Toy Adult Mens Toy (Upgrade.Stroijobs.Com)
Siktiğimin dolandırıcıları, sizi bulacağım ve bu tür dolandırıcılıklardan hiçbirini yapmazsanız sizi çok zor becereceğim Yetişkin Porno
Robot Robotic Vacuum Cleaners Tools To Ease Your
Everyday Lifethe Only Robot Robotic Vacuum Cleaners Trick Every Individual Should Be Able To
robot Robotic vacuum Cleaners
5 Killer Quora Answers To Veleco Electric Scooter Veleco Electric Scooter
You’ll Be Unable To Guess High Bed With Slide’s Tricks High Bed With Slide
We are the best and most trusted money-making site you can visit right now. With the best article methods, you’ll get maximum results.
SAJITOTO
Excellent pieces. Keep writing such kind of info on your page.
Im really impressed by your site.
Hi there, You have done an incredible job. I will certainly digg it and in my view suggest to my friends.
I am sure they will be benefited from this website.
Hi there excellent blog! Does running a blog like this require
a massive amount work? I have very little knowledge of coding but I had been hoping to start my own blog in the near future.
Anyway, should you have any ideas or tips for new blog owners please share.
I understand this is off topic nevertheless I simply needed to
ask. Cheers!
Enjoyed every bit of your post.Really looking forward to read more. Fantastic.
Link exchange is nothing else but it is simply placing the other person’s website link on your page at proper place and other
person will also do similar for you.
Nice post. I used to be checking continuously this weblog and I’m inspired!
Extremely helpful info specifically the remaining section 🙂 I care
for such info much. I was seeking this certain info for a long time.
Thanks and good luck.
I truly appreciate this blog post. Really Cool.
Muchos Gracias for your blog.Thanks Again. Really Great.
Thanks a lot for the blog article.Thanks Again. Really Great.
Thank you for your blog.
I cannot thank you enough for the article post.
A big thank you for your blog post.Much thanks again.
Hello There. I found your blog using msn. This is a really
well written article. I will be sure to bookmark it and return to read more of your useful
info. Thanks for the post. I’ll certainly comeback.
This piece of writing is truly a nice one it assists new web visitors, who are wishing in favor of blogging.
What’s up to every body, it’s my first pay a visit
of this weblog; this web site contains remarkable and genuinely excellent
stuff in support of readers.
I pay a visit daily a few web pages and websites to read articles,
but this web site gives quality based writing.
Yes! Finally something about http://www.riveredgeonline.com.
Incredible! This blog looks just like my old one!
It’s on a entirely different subject but it has
pretty much the same page layout and design. Wonderful choice of colors!
I do not even know how I ended up here, but I thought this post was great.
I don’t know who you are but certainly you’re going to a famous blogger if you aren’t already 😉 Cheers!
This excellent website truly has all the information and facts I wanted concerning
this subject and didn’t know who to ask.
I pay a quick visit day-to-day some websites and sites to read content, however
this webpage gives feature based posts.
I don’t even know the way I finished up here, but I believed this put up was great.
I don’t recognise who you are however definitely you’re going to
a well-known blogger for those who aren’t already.
Cheers!
Hi, I check your blogs like every week. Your humoristic style is awesome, keep up
the good work!
I just like the valuable info you supply on your articles.
I will bookmark your weblog and check again right here
regularly. I’m slightly sure I’ll be told plenty of new stuff proper here!
Good luck for the next!
SAJITOTO Slot
This is really interesting, You’re a very skilled blogger.
I’ve joined your feed and look forward to seeking more of your fantastic post.
Also, I have shared your website in my social networks!
Hello to every single one, it’s really a good for me to visit this site, it consists of
valuable Information.
Hi! I know this is kind of off topic but I was
wondering which blog platform are you using for this site? I’m
getting tired of WordPress because I’ve had issues with hackers and I’m looking
at options for another platform. I would be fantastic if you could point me
in the direction of a good platform.
It’s a shame you don’t have a donate button! I’d
certainly donate to this brilliant blog! I guess for now i’ll settle for bookmarking and adding
your RSS feed to my Google account. I look forward to fresh updates and will share this blog with my Facebook group.
Chat soon!
Thank you a lot for sharing this with all of us you really
realize what you’re talking about! Bookmarked. Kindly also seek advice from my web
site =). We may have a link trade agreement between us
Google’s best recommended site, don’t choose the wrong one, guys.
https://sajitoto-slot.com/
I blog quite often and I truly appreciate your content.
This great article has truly peaked my interest.
I’m going to book mark your blog and keep checking for new details about once per week.
I subscribed to your RSS feed too.
htxcjhhkp juoxx oisovzj souk nxmgvoexshfechb
Asking questions are truly fastidious thing if you are not understanding anything completely, but this piece of writing provides fastidious
understanding even.
Your style is very unique compared to other people I’ve read stuff from.
Thank you for posting when you have the opportunity, Guess I’ll just book mark this page.
bookmarked!!, I really like your site!
Undeniably believe that that you said. Your favourite justification seemed to be
at the net the easiest factor to consider of. I say to you, I definitely get irked whilst people consider worries that they just don’t recognize about.
You managed to hit the nail upon the highest as smartly
as outlined out the entire thing without having side effect , folks could
take a signal. Will likely be back to get more. Thank you
I really like what you guys tend to be up too.
This sort of clever work and coverage! Keep up the good
works guys I’ve you guys to blogroll.
Magnificent goods from you, man. I’ve take into accout your stuff previous to and you are
just extremely excellent. I really like what you’ve
received here, really like what you are stating and the best way through which you are saying it.
You’re making it enjoyable and you continue to care for to keep
it sensible. I can’t wait to learn much more from you.
That is actually a wonderful web site.
I¦ve read some excellent stuff here. Certainly price bookmarking for revisiting. I surprise how much attempt you place to make this type of fantastic informative website.
Thanks for sharing your thoughts about bokep indo.
Regards
Greate pieces. Keep writing such kind of information on your site.
Im really impressed by it.
Hey there, You have performed a fantastic job. I will certainly digg it
and in my view recommend to my friends. I’m sure they will be benefited from this
website.
What’s up mates, how is the whole thing, and what you desire
to say on the topic of this paragraph, in my
view its genuinely awesome in support of me.
Wow, superb blog layout! How long have you been blogging for?
you make blogging look easy. The overall look of your web site is excellent, as well as
the content!
This is my first time pay a quick visit at here and i am really
pleassant to read all at alone place.
Saji toto is one of the best and most trusted slot sites that you can play using the official alternative link right now.
We absolutely love your blog and find a lot of your post’s to be
exactly what I’m looking for. can you offer guest writers to write content for
you? I wouldn’t mind composing a post or elaborating on most of the subjects you write about here.
Again, awesome weblog!
you’re truly a excellent webmaster. The website
loading velocity is incredible. It seems that you are doing any distinctive trick.
Furthermore, The contents are masterwork. you’ve done a great job
on this subject!
Hello there, just became aware of your blog through Google, and found that it is really
informative. I’m gonna watch out for brussels.
I will be grateful if you continue this in future. A lot
of people will be benefited from your writing.
Cheers!
Hey there just wanted to give you a brief heads up and let you know a few of the images aren’t loading
properly. I’m not sure why but I think its a linking
issue. I’ve tried it in two different internet browsers and both show the same
results.
Whats up are using WordPress for your site platform?
I’m new to the blog world but I’m trying to get started
and set up my own. Do you need any html coding expertise to make
your own blog? Any help would be greatly appreciated!
Hi there everyone, it’s my first visit at this website,
and article is really fruitful in support of me, keep up posting such articles or
reviews.
I’ve been browsing online more than 4 hours today,
yet I never found any interesting article like yours.
It is pretty worth enough for me. In my view, if all
web owners and bloggers made good content as you did, the internet
will be a lot more useful than ever before.
The best and most trusted recommendation site is only with us, visit immediately.
Link Alternatif SAJITOTO
Do you mind if I quote a few of your articles
as long as I provide credit and sources back to your
weblog? My website is in the very same area of interest as yours and my visitors would definitely benefit
from some of the information you provide here.
Please let me know if this okay with you. Regards!
Why people still use to read news papers when in this technological world the whole
thing is available on web?
I do trust all the ideas you have presented for your post.
They’re really convincing and can definitely work.
Nonetheless, the posts are very brief for newbies.
May you please lengthen them a little from subsequent time?
Thanks for the post.
Hello, Neat post. There’s a problem with your web site in web explorer, might check this?
IE nonetheless is the marketplace leader and a big element of people will omit your great writing because of this problem.
Every weekend i used to go to see this web page,
for the reason that i want enjoyment, as this this website conations truly pleasant funny stuff too.
Awesome post.
For the reason that the admin of this web site is working,
no doubt very shortly it will be well-known, due to its feature contents.
We stumbled over here different web address and thought I might check things out.
I like what I see so now i’m following you. Look forward to checking out your
web page again.
Your style is really unique in comparison to other people I’ve read stuff
from. Thank you for posting when you have the opportunity,
Guess I will just book mark this blog.
My spouse and I stumbled over here by a different web page and
thought I might as well check things out. I like what I see so i am
just following you. Look forward to exploring your web page yet again.
Hi there! I could have sworn I’ve been to this blog before but
after browsing through some of the post I realized it’s new to me.
Anyways, I’m definitely glad I found it and I’ll be bookmarking
and checking back frequently!
You made some really good points there. I checked on the net for more info about the issue and found most
individuals will go along with your views on this website.
Everything is very open with a precise description of the
challenges. It was definitely informative. Your website is very useful.
Thanks for sharing!
Wow, marvelous blog format! How lengthy have you been running a blog for?
you made blogging look easy. The overall glance of your site is magnificent, as well as the content!
It’s actually a nice and helpful piece of information. I’m
happy that you just shared this helpful information with us.
Please stay us up to date like this. Thanks for sharing.
Situs terpercaya dan amanah jaminan pendatang baru auto maxwin
JIWA4D Login
Situs terpercaya dan amanah jaminan pendatang baru auto maxwin
If you want to take a good deal from this article then you
have to apply such strategies to your won blog.
When I initially commented I appear to have clicked
on the -Notify me when new comments are added- checkbox and from now on every time a
comment is added I receive four emails with the exact same comment.
There has to be an easy method you can remove me from that service?
Appreciate it!
Definitely consider that that you stated. Your favorite reason seemed to be on the web the easiest thing to take note of.
I say to you, I definitely get irked even as people consider concerns that they just don’t know about.
You managed to hit the nail upon the top and also outlined out the
entire thing without having side-effects , other people could take a signal.
Will probably be back to get more. Thank you
Hi there! This blog post could not be written any better!
Reading through this article reminds me of my previous roommate!
He always kept preaching about this. I will send this post
to him. Pretty sure he’ll have a great read. Thank you for sharing!
I always used to read piece of writing in news papers but now as I am a user of internet thus from now I am using net for articles, thanks to web.
Hello there! This post could not be written any better!
Looking at this article reminds me of my previous roommate!
He continually kept preaching about this. I am going to send this article to him.
Pretty sure he’ll have a very good read. Thanks for sharing!
Woah! I’m really enjoying the template/theme
of this blog. It’s simple, yet effective. A lot of times it’s challenging to get that “perfect balance” between user friendliness and
appearance. I must say you have done a amazing job with
this. In addition, the blog loads very fast for me on Internet explorer.
Superb Blog!
This is a topic that is close to my heart… Thank you! Exactly where are your contact details though?
Hello! I know this is kinda off topic but I’d figured
I’d ask. Would you be interested in exchanging links
or maybe guest writing a blog article or vice-versa? My site discusses a
lot of the same subjects as yours and I feel we could greatly benefit
from each other. If you happen to be interested feel free to shoot me an email.
I look forward to hearing from you! Superb blog by the way!
I’m really impressed with your writing skills and also
with the layout on your weblog. Is this a paid
theme or did you modify it yourself? Anyway keep up the excellent quality
writing, it is rare to see a great blog like this one today.
That is a good tip especially to those fresh to the blogosphere.
Brief but very precise information… Many thanks for sharing this one.
A must read post!
Paragraph writing is also a fun, if you know then you can write
if not it is difficult to write.
Excellent post. I was checking continuously this blog and I am impressed!
Extremely useful info specially the last part 🙂 I care for such information much.
I was looking for this certain information for a long
time. Thank you and good luck.
Hurrah, that’s what I was looking for, what a material!
existing here at this web site, thanks admin of this web page.
I always emailed this webpage post page to all my friends, because if like to read it then my contacts will too.
Wonderful goods from you, man. I have understand your stuff previous to and you are just
extremely excellent. I really like what you’ve acquired here, really like what you
are stating and the way in which you say it. You make it entertaining and you still take care of to keep it wise.
I cant wait to read much more from you. This is actually a terrific website.
Heya just wanted to give you a quick heads up and
let you know a few of the pictures aren’t loading correctly.
I’m not sure why but I think its a linking issue. I’ve tried it in two different internet browsers and both show
the same outcome.
I am truly grateful to the holder of this website who has shared this impressive article at at
this place.
I think the admin of this web site is in fact working hard for his web site,
because here every stuff is quality based material.
Hello to every one, for the reason that I am actually eager
of reading this weblog’s post to be updated daily.
It contains nice data.
Since the admin of this web site is working, no uncertainty very shortly it will be well-known, due to its quality contents.
Quality posts is the important to be a focus for the people to pay a visit the web site,
that’s what this web site is providing.
This post offers clear idea in favor of the new visitors of blogging, that really how to do blogging and site-building.
Hello to every body, it’s my first pay a visit of this weblog; this blog carries awesome and
actually fine material in support of readers.
Hey just wanted to give you a quick heads up. The words in your post seem to be running off the screen in Safari.
I’m not sure if this is a formatting issue or something to do with internet browser
compatibility but I figured I’d post to let you know. The design look great though!
Hope you get the problem resolved soon. Cheers
Situs SAJITOTO: Aplikasi Canggih Untuk Korban Encana Alam
Situs SAJITOTO: Aplikasi Canggih Untuk Korban Encana Alam
SAJITOTO
Good post. I learn something totally new and challenging
on websites I stumbleupon everyday. It will always be exciting to read through content from other
authors and use a little something from other sites.
I got this web page from my buddy who told me regarding
this web page and at the moment this time I am browsing this web site
and reading very informative articles or reviews at this
time.
Hello friends, how is everything, and what you want to say concerning
this post, in my view its in fact amazing in favor of me.
Do you have any video of that? I’d want to find out some
additional information.
Everyone loves what you guys are up too. Such clever work and coverage!
Keep up the good works guys I’ve included you guys to our blogroll.
I’m not sure exactly why but this website is loading very slow for me.
Is anyone else having this issue or is it a issue on my end?
I’ll check back later on and see if the problem still
exists.
Hmm it seems like your site ate my first comment (it was extremely long) so I guess I’ll just sum it up what I submitted and say,
I’m thoroughly enjoying your blog. I as well am an aspiring blog writer but I’m still
new to the whole thing. Do you have any tips for rookie blog writers?
I’d certainly appreciate it.
Greetings! This is my first comment here so I just wanted to give
a quick shout out and tell you I genuinely enjoy reading your articles.
Can you suggest any other blogs/websites/forums that
go over the same subjects? Thank you so much!
This is my first time pay a quick visit at here and i am actually impressed
to read everthing at one place.
Hi it’s me, I am also visiting this website regularly, this web
site is genuinely nice and the people are truly sharing good thoughts.
Great post. I was checking continuously this blog and I am impressed!
Extremely helpful info specifically the last part 🙂 I care for such information a lot.
I was looking for this certain information for a long time.
Thank you and best of luck.
This is the right site for everyone who hopes to
understand this topic. You understand so much its almost
tough to argue with you (not that I actually will need to…HaHa).
You definitely put a new spin on a subject that’s been discussed for ages.
Excellent stuff, just great!
When I originally left a comment I seem to have clicked on the -Notify me
when new comments are added- checkbox and
now each time a comment is added I recieve 4 emails with
the same comment. Perhaps there is an easy method you can remove me from that service?
Thanks!
Hi friends, nice post and fastidious arguments
commented at this place, I am really enjoying by these.
Admiring the dedication you put into your blog and detailed information you offer.
It’s nice to come across a blog every once in a while that isn’t the same out of date rehashed information. Wonderful read!
I’ve bookmarked your site and I’m adding your RSS feeds to my Google account.
I delight in, result in I found just what I was looking for.
You have ended my four day long hunt! God Bless you man. Have a nice
day. Bye
Whoa! This blog looks just like my old one! It’s on a entirely different
subject but it has pretty much the same layout and design. Wonderful choice of colors!
Hello there, You’ve done a great job. I will definitely digg it
and personally suggest to my friends. I am confident they’ll be benefited from this web
site.
Excellent site you have got here.. It’s hard to find good quality writing like yours these days.
I truly appreciate people like you! Take care!!
I think the admin of this web site is really working hard in support of his web page, for the reason that here
every material is quality based material.
I simply could not depart your site prior to suggesting that I really loved the standard
information a person supply for your guests? Is going to be back often to
inspect new posts
Amazing! Its really awesome paragraph, I have got much clear idea concerning from this
paragraph.
I love what you guys tend to be up too. Such clever work and reporting!
Keep up the excellent works guys I’ve incorporated you guys
to blogroll.
Saji toto is one of the best and most trusted slot sites that you can play using the official alternative link right now.
SAJITOTO
Saji toto is one of the best and most trusted slot sites that you can play using the official alternative link right now.
SAJITOTO
Appreciate the recommendation. Will try it out.
Yes! Finally something about stake.
Hello, I enjoy reading through your article. I like to write a little comment
to support you.
I all the time used to read paragraph in news papers but
now as I am a user of internet therefore from now I am using net for articles, thanks to web.
Wonderful article! This is the type of info that
should be shared around the internet. Shame on the search engines for now not positioning this submit higher!
Come on over and consult with my website . Thanks =)
Way cool! Some extremely valid points! I appreciate you penning this write-up
plus the rest of the site is also very good.
Heya! I just wanted to ask if you ever have any trouble with hackers?
My last blog (wordpress) was hacked and I ended up losing
months of hard work due to no backup. Do you have any methods to prevent hackers?
Hello to all, how is everything, I think every one is getting
more from this web site, and your views are nice for new visitors.
Thanks on your marvelous posting! I genuinely enjoyed reading it, you will be a great author.
I will ensure that I bookmark your blog and may come back down the road.
I want to encourage you to definitely continue your great
job, have a nice evening!
Hey there! I’ve been following your blog for some time now and finally
got the courage to go ahead and give you a shout out from Atascocita Texas!
Just wanted to mention keep up the great job!
Hi! I’ve been following your blog for some time now
and finally got the courage to go ahead and give you a shout out from Houston Texas!
Just wanted to say keep up the fantastic work!
you are really a good webmaster. The website loading speed is amazing. It seems that you are doing any unique trick. In addition, The contents are masterpiece. you’ve done a wonderful job on this topic!
If some one desires expert view regarding blogging and site-building then i recommend him/her to go to see this website, Keep up the fastidious work.
What a material of un-ambiguity and preserveness of valuable familiarity
regarding unexpected feelings.
Have you ever considered about including a little bit more than just your articles?
I mean, what you say is fundamental and everything. But imagine if you added some great images
or video clips to give your posts more, “pop”! Your content is excellent but
with pics and video clips, this site could certainly
be one of the most beneficial in its field. Very good blog!
Hi there would you mind sharing which blog platform you’re working with?
I’m planning to start my own blog soon but I’m having
a hard time selecting between BlogEngine/Wordpress/B2evolution and Drupal.
The reason I ask is because your layout seems
different then most blogs and I’m looking for something completely unique.
P.S Apologies for being off-topic but I had to ask!
I’m not sure why but this site is loading very slow for
me. Is anyone else having this problem or is it a issue on my end?
I’ll check back later and see if the problem still exists.
Heya i’m for the first time here. I came across this board
and I find It truly useful & it helped me out much.
I hope to give something back and help others like you helped me.
You are so interesting! I do not think I have read through a single thing like this
before. So great to find someone with a few genuine thoughts on this issue.
Seriously.. thanks for starting this up. This
website is one thing that is required on the internet, someone with a little originality!
What’s up to all, the contents present at this site are genuinely amazing
for people knowledge, well, keep up the good work fellows.
Hi! I’ve been reading your site for a long time now and finally got the courage to go ahead
and give you a shout out from Atascocita Texas! Just wanted
to mention keep up the excellent work!
I’m truly enjoying the design and layout of your blog.
It’s a very easy on the eyes which makes it much more enjoyable for me to come here and visit more often. Did you hire out a developer to create your theme?
Excellent work!
I am really inspired along with your writing abilities as neatly as with the format in your blog.
Is this a paid subject or did you customize it your self?
Either way stay up the excellent quality writing, it
is rare to look a nice weblog like this one today..
Right now it appears like WordPress is the best blogging platform
available right now. (from what I’ve read) Is that what you
are using on your blog?
Hello I am so happy I found your site, I really found you by mistake, while I was searching on Digg for something else, Regardless I am here now and would just like to say thanks a lot for a tremendous post and a all round interesting blog (I also love the theme/design), I don’t have time to read it all at the moment but I have saved it and also included your RSS feeds, so when I have time I will be back to read much more, Please
do keep up the fantastic work.
Hi there I am so grateful I found your web site, I really found you by
error, while I was researching on Askjeeve for something else,
Anyhow I am here now and would just like to say thank
you for a fantastic post and a all round enjoyable blog (I also love the theme/design), I don’t have
time to go through it all at the moment but I have bookmarked it and
also added in your RSS feeds, so when I have time
I will be back to read a great deal more, Please do keep up the great work.
I love it when people get together and share views.
Great blog, keep it up!
The SAJITOTO is a place to compete for the prestige of the nnewest space adventure game.
SAJITOTO
The SAJITOTO is a place to compete for the prestige of the nnewest space adventure game.
SAJITOTO
Very good information. Lucky me I came across
your blog by chance (stumbleupon). I’ve book marked it for later!
Attractive element of content. I simply stumbled upon your
website and in accession capital to claim that I acquire
in fact enjoyed account your weblog posts. Anyway I’ll be subscribing in your
feeds and even I success you access persistently fast.
Hi to every one, the contents present at this site are really amazing for people experience, well, keep up the good work fellows.
This is very fascinating, You are an overly professional blogger.
I’ve joined your feed and stay up for in search of extra of your fantastic
post. Additionally, I have shared your website in my social networks
I am really impressed with your writing skills as well
as with the layout on your weblog. Is this a paid theme or did you modify it yourself?
Anyway keep up the nice quality writing, it’s rare to see a great blog
like this one nowadays.
Somebody essentially lend a hand to make critically
articles I might state. This is the first time I frequented your website page and up
to now? I surprised with the research you made to make
this actual post amazing. Great job!
Howdy! Someone in my Myspace group shared this website with us so I came to take a
look. I’m definitely loving the information. I’m
book-marking and will be tweeting this to my followers!
Fantastic blog and wonderful design.
always i used to read smaller posts which also clear their
motive, and that is also happening with
this post which I am reading at this place.
Hello! I could have sworn I’ve been to this site before
but after going through many of the posts I realized it’s new to me.
Anyways, I’m definitely happy I found it and I’ll be book-marking it
and checking back regularly!
I like the valuable information you provide in your articles.
I’ll bookmark your weblog and check again here frequently.
I am quite sure I’ll learn many new stuff right here!
Best of luck for the next!
Does your site have a contact page? I’m
having problems locating it but, I’d like to send you an e-mail.
I’ve got some recommendations for your blog you might be interested in hearing.
Either way, great blog and I look forward to seeing it improve over time.
Hello there! This blog post couldn’t be written much better!
Going through this article reminds me of my previous roommate!
He constantly kept preaching about this. I am going to forward this post to him.
Pretty sure he’ll have a very good read. Thanks for sharing!
Appreciating the dedication you put into your website and
detailed information you offer. It’s awesome to come across a blog every
once in a while that isn’t the same unwanted rehashed material.
Fantastic read! I’ve bookmarked your site and I’m adding your RSS feeds to my Google account.
Wow, marvelous blog layout! How long have you been blogging for?
you make blogging look easy. The overall
look of your web site is fantastic, as well as the content!
It’s awesome to pay a visit this web site and reading the views of all friends regarding
this article, while I am also eager of getting experience.
Hi there, I enjoy reading all of your article. I wanted to write a little comment to support you.
I’ve read a few good stuff here. Definitely value bookmarking for revisiting.
I wonder how much attempt you place to create
any such excellent informative website.
I’m curious to find out what blog platform you have been working with?
I’m having some small security problems with my latest
blog and I would like to find something more safe. Do you have any recommendations?
I love your blog.. very nice colors & theme. Did you make this
website yourself or did you hire someone to do it for you?
Plz answer back as I’m looking to design my own blog
and would like to find out where u got this from. many thanks
An impressive share! I’ve just forwarded this onto a co-worker who has been doing a
little homework on this. And he in fact bought me breakfast because I stumbled upon it for him…
lol. So allow me to reword this…. Thanks for the meal!!
But yeah, thanx for spending time to talk about this matter here on your
internet site.
Woah! I’m really digging the template/theme of this blog.
It’s simple, yet effective. A lot of times it’s difficult to get that
“perfect balance” between superb usability and visual appearance.
I must say you’ve done a amazing job with this. Additionally, the
blog loads very fast for me on Safari. Excellent Blog!
Good web site you have here.. It’s difficult to find excellent writing like yours these days.
I really appreciate individuals like you! Take care!!
I all the time used to read post in news papers but now as I am a user
of web therefore from now I am using net for articles,
thanks to web.
I am extremely inspired along with your writing talents as neatly as with
the structure to your blog. Is this a paid subject
matter or did you modify it yourself? Anyway stay up the excellent quality writing, it
is uncommon to peer a nice weblog like this one these days..
Yes! Finally something about post surgery scar treatment.
Hi! I understand this is somewhat off-topic however
I needed to ask. Does building a well-established website such as
yours require a lot of work? I am brand new to writing a blog however I do write in my journal every day.
I’d like to start a blog so I will be able to share my own experience and feelings online.
Please let me know if you have any recommendations or tips for new aspiring bloggers.
Thankyou!
Yes! Finally someone writes about rundpools set.
Do you have a spam problem on this site; I also am a blogger,
and I was curious about your situation; many of us have created some nice procedures and we are looking
to swap techniques with others, please shoot me an e-mail if interested.
Hello! I know this is somewhat off topic but I was wondering which blog platform
are you using for this site? I’m getting sick and tired of WordPress
because I’ve had issues with hackers and I’m looking at options
for another platform. I would be awesome if you could point me in the direction of a
good platform.
This web site truly has all of the information I needed
concerning this subject and didn’t know who to ask.
Good day I am so thrilled I found your website, I really found
you by mistake, while I was searching on Askjeeve
for something else, Anyways I am here now and would just
like to say thanks for a incredible post and
a all round entertaining blog (I also love the theme/design), I don’t have time to
read through it all at the minute but I have book-marked it and also included your RSS feeds, so
when I have time I will be back to read much more, Please do keep up the excellent jo.
An interesting discussion is definitely worth comment.
There’s no doubt that that you need to publish more on this subject matter, it might not be
a taboo matter but typically folks don’t discuss these subjects.
To the next! Best wishes!!
Have you ever thought about adding a little bit
more than just your articles? I mean, what you say is fundamental and all.
But just imagine if you added some great graphics or videos to give your posts
more, “pop”! Your content is excellent but with images and video clips, this website could undeniably be one of
the best in its field. Awesome blog!
Right now it looks like Drupal is the best blogging platform out there right now.
(from what I’ve read) Is that what you’re using on your blog?
Woah! I’m really enjoying the template/theme of this blog.
It’s simple, yet effective. A lot of times it’s challenging to get that “perfect balance” between usability and visual
appearance. I must say you have done a very good
job with this. In addition, the blog loads very fast for me
on Safari. Exceptional Blog!
What’s up, yes this piece of writing is actually pleasant and I have learned
lot of things from it regarding blogging. thanks.
An outstanding share! I have just forwarded this onto a coworker who was conducting a
little homework on this. And he actually bought me dinner because I stumbled
upon it for him… lol. So allow me to reword this….
Thanks for the meal!! But yeah, thanks for spending
the time to talk about this subject here on your site.
Hmm it appears like your blog ate my first comment (it was extremely long) so I guess
I’ll just sum it up what I wrote and say, I’m
thoroughly enjoying your blog. I as well am
an aspiring blog blogger but I’m still new to everything.
Do you have any tips and hints for beginner blog writers?
I’d definitely appreciate it.
Remarkable! Its really amazing piece of writing, I have got
much clear idea regarding from this post.
SAJITOTO is the best income website of all time that you can visit now.
SAJITOTO is the best income website of all time that you can visit now.
classic game full of challenges only here guys.
SAJI TOTO Ink
classic game full of challenges only here guys.
SAJI TOTO Ink
The best online game trailer that you must try this time.
SANJITOTO
Just want to say your article is as astounding.
The clearness in your post is simply nice and that i can assume you
are an expert in this subject. Well together with your permission allow me to
grasp your RSS feed to stay updated with drawing close post.
Thank you one million and please keep up the gratifying work.
I simply could not go away your web site before
suggesting that I actually enjoyed the usual information an individual provide in your guests?
Is going to be back ceaselessly to inspect new posts
You really make it seem so easy with your presentation but I find this topic to be really something that I think I would never understand. It seems too complicated and very broad for me. I am looking forward for your next post, I’ll try to get the hang of it!
Very good site you have here but I was curious if you knew
of any community forums that cover the same topics discussed in this article?
I’d really like to be a part of community where I can get comments from other experienced individuals that share the same interest.
If you have any suggestions, please let me know.
Cheers!
I’ll immediately snatch your rss feed as I can’t to find your
e-mail subscription hyperlink or e-newsletter service.
Do you have any? Kindly permit me recognize in order that I
may just subscribe. Thanks.
Thank you, Regard Telkom University
Thank you for information!
Hey there, You’ve done a great job. I will definitely digg it and personally suggest
to my friends. I am sure they will be benefited from this
website.
I could not resist commenting. Well written!
Im Gegensatz zu einigen Wettbewerbern, bei denen Freispiele nur für bestimmte oder ältere Spiele verfügbar sind, enthält das Paket von Mystake oft sowohl Cash-Boosts als auch Freispiele. Einige Werbeguthaben oder Freispiele können mit unterschiedlichen Rollover-Multiplikatoren ausgestattet sein. Mit einer VIP-Mitgliedschaft können Sie schneller Geld abheben und Hilfe von einem persönlichen Manager erhalten, was Ihr Spielerlebnis noch besser macht.
Die richtigen lokalen Begriffe werden automatisch angezeigt, wenn Sie die deutsche Benutzeroberfläche verwenden. Um die Dinge klar und einfach zu halten, teilen wir aktive Codes an mehr als einem Ort. Für die meisten Mystake Casino Codes reicht eine Mindesteinzahlung von €20 aus, es sei denn, die Aktion sagt ein höheres Limit. Mit den Casino-Einstellungen können Sie die Benachrichtigungen steuern, die Sie für zeitlich begrenzte Drops und Renneinladungen erhalten. Für jedes €1, das Sie auf bestimmte Slots setzen, erhalten Sie einen Punkt. Bevor Sie beginnen, schauen Sie sich den Beitragshinweis auf der Promo-Karte oder die Seite mit den Bonusbedingungen in der Casino-Lobby an, um sicherzustellen, dass Sie abgesichert sind.
Deswegen verwenden wir Encryption Methoden, um dich und deine Daten zu schützen. Wenn Sie nach der MyStake App gesucht haben und nichts Passendes finden konnten, machen Sie sich keine Sorgen. Wähle einfach deine bevorzugte Zahlungsmethode und wir erledigen den Rest. Es war noch nie so einfach, dein Guthaben aufzuladen oder dir deine Gewinne auszahlen zu lassen.
References:
https://online-spielhallen.de/kings-casino-erfahrungen-umfassender-spielerbericht/
Keep this going please, great job!
Generally I don’t learn article on blogs, but I would like to say that this write-up very forced me to take a look at and
do so! Your writing taste has been amazed me.
Thanks, quite nice article.
Good day! I simply want to offer you a huge thumbs up for
your great information you have here on this post.
I am returning to your blog for more soon.
Spot on with this write-up, I honestly feel this
website needs far more attention. I’ll probably be back again to read more, thanks for the advice!
Have you ever considered publishing an e-book or guest authoring on other blogs?
I have a blog centered on the same information you discuss and would really like to have you share some stories/information. I know my
subscribers would enjoy your work. If you’re even remotely interested, feel free to
send me an e-mail.
Great blog! Is your theme custom made or did you download it
from somewhere? A theme like yours with a few simple adjustements would really make my
blog stand out. Please let me know where you got your design. Appreciate it
Thankfulness to my father who stated to me regarding this website,
this blog is really amazing.
Hi, I think your website might be having browser compatibility issues.
When I look at your blog in Firefox, it looks fine but when opening in Internet Explorer, it
has some overlapping. I just wanted to give you a quick heads up!
Other then that, awesome blog!
Do you have a spam issue on this site; I also am a blogger, and I was wanting to
know your situation; we have created some nice methods and we are looking to trade methods with other folks, be sure to shoot me an email if interested.
Right now it seems like Drupal is the best blogging
platform available right now. (from what I’ve read) Is that what you’re using on your blog?
Can I simply say what a comfort to find someone who actually knows what they’re
discussing on the web. You certainly know how to bring an issue to light and make
it important. More and more people ought to check this
out and understand this side of your story. I can’t believe you’re not more popular given that you most certainly possess the gift.
All the games at this online casino are powered by the top-tier software provider, Real Time Gaming. Video Poker games have gradually become one of the most popular games ever played online. Players certainly won’t be disappointed with the wide but selective collection of Video Poker games available at this online casino. There’s also a separate section where players have the advantage of adding their favourite games under the same head for easy accessibility.
The app is optimized for mobile devices, ensuring that gameplay is smooth, responsive, and fun. We want to make sure that you feel supported and confident while enjoying your gaming experience with us. Whether you’re playing on your desktop or mobile device, you can reach out to our support team at any time for prompt assistance. We understand that players want a smooth experience, and we make sure that all your payments are reliable, secure, and convenient.
The welcome package gives players match bonuses of up to $1000. Fair Go Casino bonuses start with a bang, with a generous Welcome Bonus offered to new players. It starts with a generous welcome bonus and continues with multiple weekly and monthly bonuses for loyal players. The game selection is on point, as are the deposit options, bonuses, and promotions.
References:
https://blackcoin.co/this-is-vegas-casino-review/
Nice answers in return of this difficulty with solid arguments and
explaining everything regarding that.
You can certainly see your expertise within the article you write.
The world hopes for even more passionate writers such as
you who aren’t afraid to say how they believe.
All the time go after your heart.
These are genuinely impressive ideas in on the topic of blogging.
You have touched some good points here. Any way keep up wrinting.
whoah this blog is magnificent i like reading your articles.
Keep up the good work! You already know, lots of persons are searching around for this info, you could help them greatly.
us poker sites that accept paypal
References:
https://externalliancerh.com/employer/best-paypal-casinos-uk-2025-casino-sites-that-accept-paypal/
online poker real money paypal
References:
https://koftc.com/bbs/board.php?bo_table=free&wr_id=21105
This really answered my problem, thank you!
online pokies australia paypal
References:
https://metagap.ro/employer/play-pokies-real-money-with-instant-withdrawal/
online australian casino paypal
References:
https://jobsonly.in/employer/best-online-casinos-in-australia-%ef%b8%8f-ranked-by-experts-2025/
Thanks for finally writing about > 医薬におけるバリデーション | お役立ち情報 < Liked it!
You could definitely see your skills in the work you write.
The sector hopes for even more passionate writers like you
who are not afraid to mention how they believe.
Always go after your heart.
I do not even know the way I ended up here, but I assumed this post was
good. I do not recognise who you’re but certainly you are going to a well-known blogger in case you are not already.
Cheers!
I was wondering if you ever considered changing the page layout of your site?
Its very well written; I love what youve got to
say. But maybe you could a little more in the way of content so people could
connect with it better. Youve got an awful lot of text for only having one or 2 images.
Maybe you could space it out better?
I was curious if you ever thought of changing the page layout of your website?
Its very well written; I love what youve got to say.
But maybe you could a little more in the way of content so people could
connect with it better. Youve got an awful lot of text for only having 1 or two images.
Maybe you could space it out better?
A motivating discussion is worth comment. There’s no doubt that that you need to write more on this subject, it may not be a taboo matter
but usually people don’t discuss such subjects. To the next!
All the best!!
I am extremely impressed with your writing talents as well as with the layout to your weblog.
Is this a paid subject matter or did you modify it yourself?
Anyway stay up the nice high quality writing, it’s rare to see a
great weblog like this one nowadays..
I think that is one of the so much significant information for me.
And i’m happy reading your article. However want to commentary on some
normal issues, The web site taste is perfect, the articles is truly excellent : D.
Good job, cheers
I think this is one of the most vital info for me.
And i’m glad reading your article. However want to commentary on some normal things,
The website taste is perfect, the articles is actually nice :
D. Good process, cheers
Pretty! This has been a really wonderful post. Thank you for supplying
this information.
Every weekend i used to pay a quick visit this site, for the reason that i
wish for enjoyment, as this this site conations really pleasant funny data too.
Someone necessarily lend a hand to make severely articles I’d state.
That is the very first time I frequented your web page and so far?
I surprised with the research you made to create this actual put up incredible.
Excellent process!
Every weekend i used to pay a visit this site, as i wish for enjoyment, since this this web site conations in fact good funny stuff
too.
Good day I am so thrilled I found your website, I really found you by accident, while I was researching on Bing for something else, Anyhow
I am here now and would just like to say thank you for a
incredible post and a all round exciting blog (I also love the theme/design), I don’t have time to go through it all
at the moment but I have saved it and also included your RSS feeds,
so when I have time I will be back to read a lot more, Please do keep up the excellent
work.
Hi! I’ve been following your web site for some time now and finally got the courage to go ahead and
give you a shout out from Houston Texas! Just wanted to tell you keep up the great work!
Thanks for ones marvelous posting! I certainly enjoyed reading it, you are a great author.
I will make sure to bookmark your blog and may come
back later on. I want to encourage yourself to continue your great posts, have a nice
evening!
Magnificent items from you, man. I’ve take into account
your stuff previous to and you’re just extremely fantastic.
I really like what you’ve bought here, really like what you’re
stating and the way in which by which you are saying it.
You make it enjoyable and you still care for to keep it smart.
I can’t wait to learn much more from you. This is really a
great website.
Hi it’s me, I am also visiting this site on a regular basis, this site is actually nice and the people are
truly sharing nice thoughts.
Very nice post. I simply stumbled upon your blog and wished to mention that I’ve truly
loved surfing around your weblog posts. In any case I will
be subscribing on your feed and I’m hoping you write again very soon!
I was recommended this website via my cousin. I’m no
longer sure whether or not this post is written via him
as no one else realize such specific approximately my difficulty.
You’re wonderful! Thanks!
This is a topic that is close to my heart…
Thank you! Where are your contact details though?
You are so awesome! I do not suppose I’ve read through something
like this before. So good to discover someone with original thoughts on this subject matter.
Seriously.. thank you for starting this up. This web site
is something that is needed on the web, someone with a little originality!
Hi there, after reading this awesome piece of writing i am too glad to share my
experience here with mates.
Since the admin of this web page is working, no question very
rapidly it will be renowned, due to its feature contents.
Have you ever considered creating an e-book or guest authoring on other websites?
I have a blog based upon on the same information you discuss and would really like
to have you share some stories/information. I know my readers would appreciate your work.
If you’re even remotely interested, feel free to send me an e mail.
Very good post! We will be linking to this particularly great article on our
website. Keep up the great writing.
obviously like your web site however you need to take a look at
the spelling on quite a few of your posts. Many of them are rife with spelling issues and I in finding it very bothersome to tell the reality nevertheless I
will surely come again again.
I’m really enjoying the theme/design of your site.
Do you ever run into any web browser compatibility issues?
A handful of my blog readers have complained about my website not working correctly in Explorer but looks great in Chrome.
Do you have any advice to help fix this issue?
Helpful information. Lucky me I discovered your web site unintentionally, and I am shocked why this accident
didn’t came about earlier! I bookmarked it.
Hello There. I discovered your weblog using msn. This is a really
neatly written article. I’ll be sure to bookmark it and return to read extra of your helpful info.
Thanks for the post. I’ll definitely comeback.
You could definitely see your enthusiasm in the article you write.
The sector hopes for more passionate writers such as
you who aren’t afraid to mention how they believe.
All the time follow your heart.
An intriguing discussion is worth comment. I do believe that you ought to write more about this subject,
it might not be a taboo matter but usually people don’t speak about these subjects.
To the next! All the best!!
Hello! I’m at work surfing around your blog from my new
iphone! Just wanted to say I love reading your blog and look forward
to all your posts! Keep up the excellent work!
Hi! I just wanted to ask if you ever have any issues with hackers?
My last blog (wordpress) was hacked and I ended up losing many months
of hard work due to no data backup. Do you have any methods to stop hackers?
Attractive portion of content. I just stumbled upon your site and in accession capital to assert that I get actually
enjoyed account your weblog posts. Any way I will be subscribing
to your feeds or even I fulfillment you get right of entry to persistently fast.
Have you ever thought about publishing an ebook or guest authoring
on other blogs? I have a blog based on the same topics you
discuss and would love to have you share some stories/information. I know my readers would value your work.
If you’re even remotely interested, feel free to send me an email.
Great goods from you, man. I have consider your
stuff previous to and you’re simply too great. I actually like what you have bought right
here, really like what you are saying and the best way wherein you
assert it. You are making it entertaining and you continue to care for to keep it sensible.
I can’t wait to read much more from you. That is really a terrific site.
Oh my goodness! an amazing article dude. Thank you However I’m experiencing challenge with ur rss . Don’t know why Unable to subscribe to it. Is there anyone getting an identical rss downside? Anyone who is aware of kindly respond. Thnkx
Today, I went to the beachfront with my kids.
I found a sea shell and gave it to my 4 year
old daughter and said “You can hear the ocean if you put this to your ear.” She
put the shell to her ear and screamed. There was a hermit crab inside and
it pinched her ear. She never wants to go back! LoL I know this is entirely off topic but I had to
tell someone!
An impressive share! I’ve just forwarded this onto a coworker who had been doing a
little research on this. And he actually bought me lunch due to the fact that
I discovered it for him… lol. So let me reword this….
Thanks for the meal!! But yeah, thanx for spending some time to discuss this topic here on your site.
Magnificent web site. Plenty of useful info here.
I’m sending it to some pals ans also sharing in delicious. And naturally, thanks in your sweat!
I’m not that much of a internet reader to be honest but your sites really nice, keep it up!
I’ll go ahead and bookmark your site to come back
later. Cheers
I’m very pleased to discover this page. I want to to thank you for ones time due
to this wonderful read!! I definitely enjoyed every little bit of it
and I have you saved to fav to look at new stuff in your web site.
It’s impressive that you are getting thoughts from this article as well as from our argument made here.
A fascinating discussion is definitely worth comment. I do believe
that you ought to write more about this topic, it might not be a taboo matter but
generally people do not talk about such topics.
To the next! Kind regards!!
I’m amazed, I have to admit. Seldom do I encounter a blog that’s equally educative
and engaging, and let me tell you, you’ve hit the nail on the head.
The issue is something which not enough men and women are speaking intelligently about.
Now i’m very happy I stumbled across this during my search for something relating to this.
Hello, this weekend is nice for me, for the
reason that this occasion i am reading this enormous educational article here at my house.
Everything is very open with a really clear description of the challenges.
It was really informative. Your site is useful. Thanks for sharing!
I don’t even know how I ended up here, but I thought this post was good.
I do not know who you are but definitely you are going to a famous blogger if you aren’t already 😉 Cheers!
I really love your website.. Pleasant colors & theme.
Did you create this site yourself? Please reply back as I’m hoping
to create my own personal site and would like to know where
you got this from or exactly what the theme is named. Kudos!
Thanks for finally talking about > 医薬におけるバリデーション | お役立ち情報 < Loved it!
Hey I know this is off topic but I was wondering if you knew of any widgets I could add to my blog that automatically tweet my newest twitter updates.
I’ve been looking for a plug-in like this for quite some time and was hoping
maybe you would have some experience with something like this.
Please let me know if you run into anything. I truly enjoy reading your blog and I look
forward to your new updates.
Thanks for sharing your info. I really appreciate your efforts and I will be waiting
for your next post thank you once again.
It’s actually a great and helpful piece of info. I’m glad that you
shared this useful information with us. Please keep us informed like this.
Thanks for sharing.
My brother recommended I might like this website. He was entirely right.
This post truly made my day. You can not imagine simply how
much time I had spent for this information! Thanks!
Hey! Would you mind if I share your blog with
my myspace group? There’s a lot of people that I think would really enjoy your content.
Please let me know. Cheers
Hmm is anyone else encountering problems with the images on this blog loading?
I’m trying to find out if its a problem on my end or if it’s the blog.
Any feed-back would be greatly appreciated.
I have been surfing on-line greater than three hours as of late, yet I never discovered
any fascinating article like yours. It’s pretty price sufficient for me.
In my view, if all website owners and bloggers
made good content as you probably did, the internet might
be much more helpful than ever before.
Helpful information. Fortunate me I found your website accidentally, and I’m surprised why this coincidence didn’t came about
in advance! I bookmarked it.
Hello, its fastidious piece of writing concerning media print,
we all know media is a wonderful source of facts.
Have you ever thought about including a little bit more than just your articles?
I mean, what you say is fundamental and all.
However think about if you added some great photos or videos to give
your posts more, “pop”! Your content is excellent but with pics and clips, this blog could certainly be one of the best in its niche.
Awesome blog!
Incredible points. Outstanding arguments. Keep up the good effort.