コンピュータ化システムの4つの種類
コンピュータ化システムは、大きく分類して4種類のカテゴリに分けられる。(図1参照)
1) プロセスコントロール(構造設備)
2) ITアプリケーション
3) ラボ(分析機器、Excel)
4) インフラストラクチャ
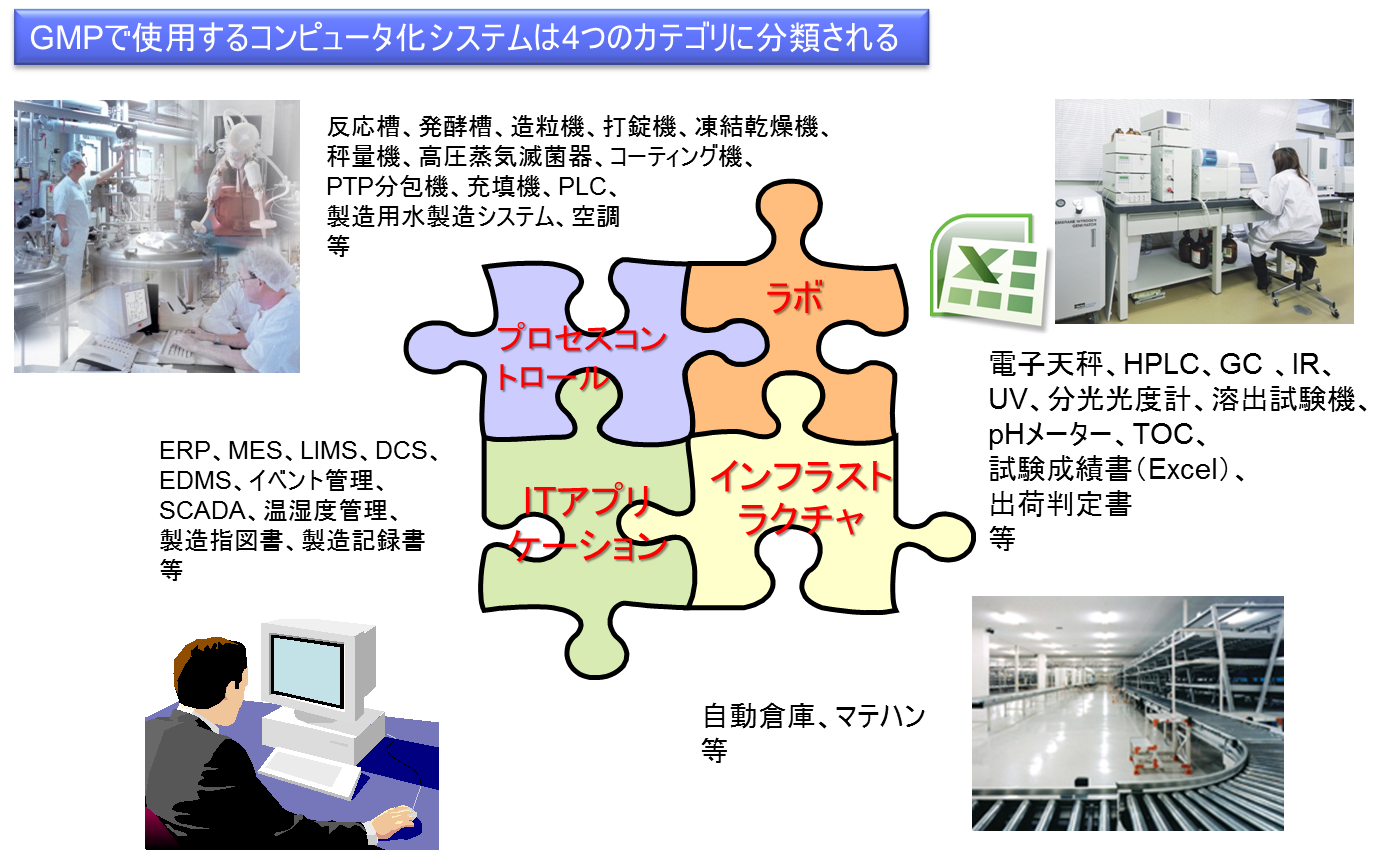
一般に、GMP関連業務においては、上記の4システムがすべて使用される。それに対して、GLP関連業務では、主にラボとITアプリケーションが使用され、GCP・GVP・GQP関連業務では、ITアプリケーションが使用される。
上記4種類のシステムは、それぞれに特徴が異なり、またバリデーションの実施方法が異なる。しかしながら、どのカテゴリにも精通した専門家はほとんどいないのが現状である。
プロセスコントロール(構造設備)
構造設備は、工場のラインに設置され、実際に原薬や製品(製剤)を生産するシステムである。
例えば、原薬工場における反応槽、発酵槽や、製剤工場における造粒機、打錠機、凍結乾燥機、秤量機、高圧蒸気滅菌器、コーティング機、PTP分包機、充填機などが相当する。
また、製造用水製造システムや空調などの支援設備(ユーティリティシステム)も構造設備に含まれる。
構造設備の品質は、製品の品質に大きく影響する。したがって、バリデーションは重要である。
「原薬GMPのガイドライン」では、プロセスバリデーションを始める前に、重要な装置及び付帯設備の適格性評価を完了することとなっている。
構造設備の特徴は、その品質が直感的に把握できることにある。すなわち、あらかじめ実際に製品を生産し、生産された製品の品質を目視チェックや分析することによって検証ができるのである。
構造設備では、PLC、ファームウェアなどの比較的小さなプログラムで制御していることが多い。
多くの構造設備は、カテゴリ3である。ただし、複雑またはユーザが変更したPLCは、カテゴリ5に分類されるが、カテゴリ3と5の境界は曖昧である。
一般に1つの構造設備は、1つの機能しかもたない。例えば、造粒機は造粒する機能、打錠機は打錠する機能である。したがって、多くの場合、構造設備では、機能仕様書は作成しない。(というよりも作成できない。)
構造設備を対象とするバリデーションを「適格性評価」と呼び、DQ(設計時適格性評価)、IQ(設備据付時適格性評価)、OQ(運転時適格性評価)、PQ(性能適格性評価)から構成される。
適格性評価は、製品品質に直接影響する要因についてのみ、設計段階でDQを、製作・施工段階でIQを、試験・検査・試運転段階でOQとPQを行うことである。
昨今は、SCADAやDCSのように、ネットワーク化され、ITアプリケーションにより中央で集中管理されている構造設備も利用されている。
旧ガイドラインではファームウェアやPLCは対象となっていなかった
平成4年2月21日に厚生省(当時)から発出された、薬監第11号「コンピュータ使用医薬品等製造所適正管理ガイドライン」(以下、旧ガイドライン)の「第2.適用範囲」には、以下の記載があった。
このガイドラインは、医薬品GMPが適用される製造所のうち、次のいずれかに該当するシステムを使用する製造所に適用する。
ただし、使用目的が限定され、そのためのプログラムがハードウェア(コンピュータにより制御される機器及び設備を含む。以下同じ。)の提供業者によって汎用機能として固定され、パラメーターを設定することによって機能が実現されるシステムを除くものとする。
この記述を読むと、旧ガイドラインでは、打錠機などの出来合いの構造設備(カテゴリ3)は除外されていたことがわかる。またファームウェアやPLC なども適用されていなかった。
新ガイドラインでは、カテゴリ3 に分類される構造設備や、それらに搭載されているPLC、ファームウェアも適用対象になったことに留意する必要がある。
図に示すとおり、新ガイドラインの別紙2にカテゴリ分類表が添付されている。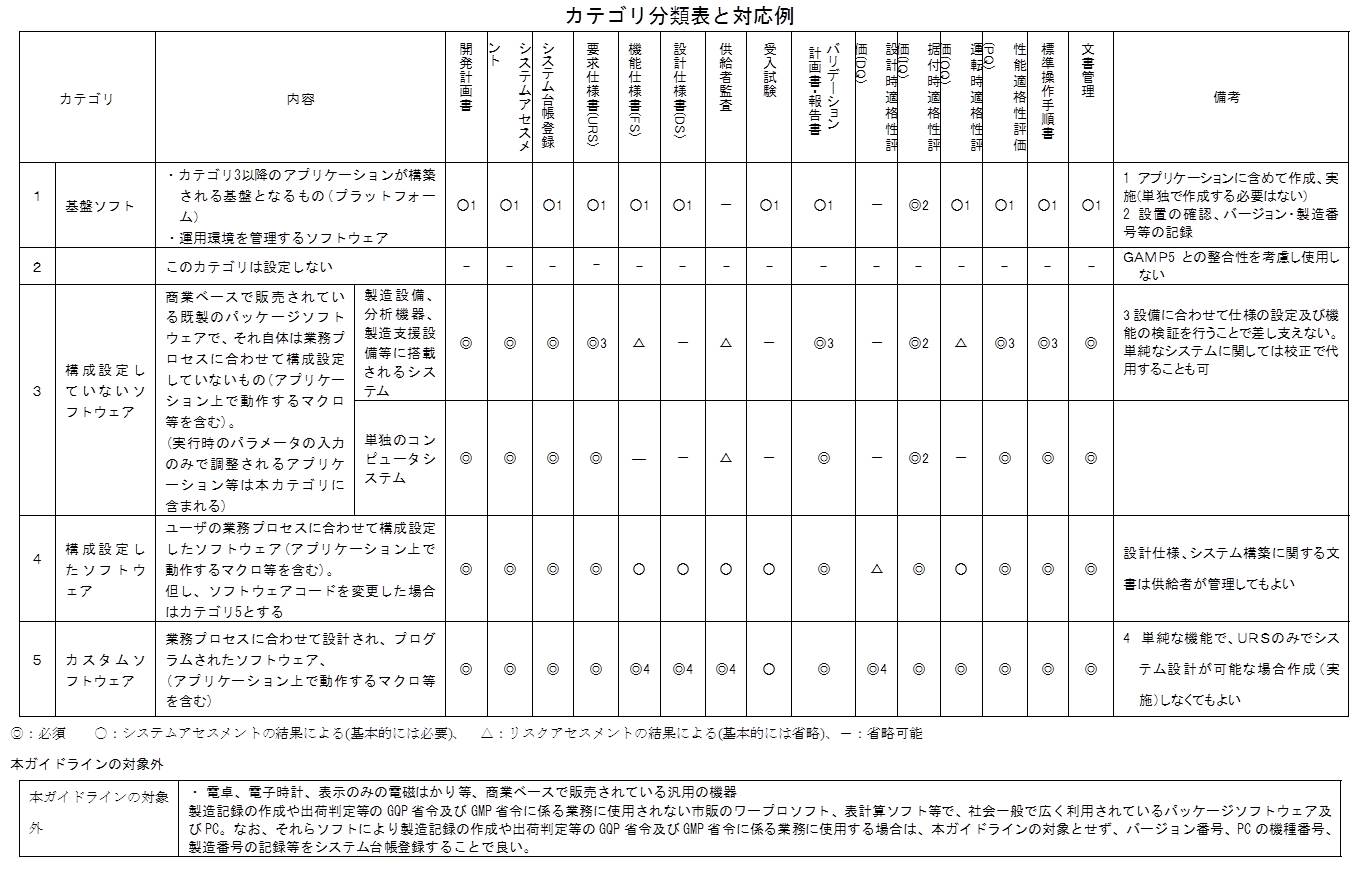
カテゴリ2は使用しないと明記されているが、これがGAMP 4では、ファームウェアやPLCであった。
実は、ファームウェアやPLCはCSV対象から外れたのではなくて、カテゴリ3の上段に移動された。
ここには「製造設備、分析機器、製造支援システム等に搭載されるシステム」とある。例えば、シーケンサー等がそれに相当するが、ファームウェアやPLCによって動作している。
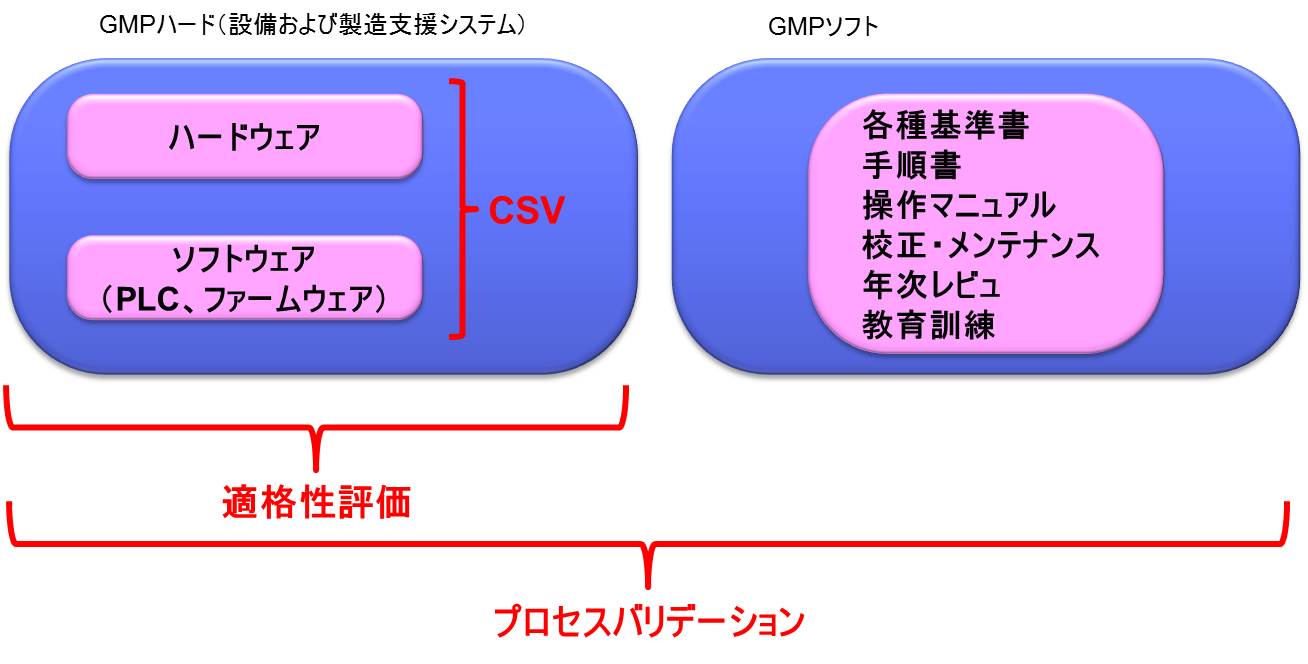
GMPにおけるハードとソフト
GMPを語る上でははずせない要素・考え方として「ハード」と「ソフト」がある。
「GMPハード」と「GMPソフト」の両者によりGMPの目的を達成するのである。
GMPハードとは、設備のことであり、例えば以下のような要件である。
- 間違いを防ぐことのできる設備・環境の製造所であること
- 衛生的な設備・環境の製造所であること
- 高い品質を保ち続けることができる設備・環境の製造所であること
一方、GMPソフトとは、ルールのことであり、以下のような要件があげられる。
- ルールを決めて文書化すること
- ルールどおりに実施し、記録を作成すること
- 定期的に見直しを行い、改善をはかること
ここで、「GMPハード」と「GMPソフト」は、「ハードウェア」、「ソフトウェア」のことではないことに注意が必要である。「ハードウェア」、「ソフトウェア」はともに「GMPハード」である。
適格性評価とは
GMPにおいて、「適格性評価」(Qualification)と呼ばれる、他の業界では使用されない特殊な用語が存在する。一般に構造設備のようなGMPハードのバリデーションについては、適格性評価を行う。
では、「適格性評価」とは、いったいどのようなものであるのだろうか。
ICH Q7を受けて平成13年に発出された「原薬GMPのガイドライン」(平成13年11月2日、薬発第1200号)の12.30において、以下のような記載がある。
12.30 プロセスバリデーションの作業を始める前に、重要な装置及び付帯設備の適格性評価を完了すること。適格性評価は、通常、以下の作業を個々に、又は組み合わせて実施する:
-設計時適格性評価(DQ):設備、装置又はシステムが目的とする用途に適切であることを確認し文書化すること。
-設備据付時適格性評価(IQ):据付け又は改良した装置又はシステムが承認を受けた設計及び製造業者の要求と整合することを確認し文書化すること。
-運転時適格性評価(OQ):据付け又は改良した装置又はシステムが予期した運転範囲で意図したように作動することを確認し文書化すること。
-性能適格性評価(PQ):設備及びそれに付随する補助装置及びシステムが、承認された製造方法及び規格に基づき、効果的かつ再現性よく機能できることを確認し文書化すること。
「プロセスバリデーションの作業を始める前に、重要な装置および付帯設備の適格性評価を完了すること」と記載のとおり、適格性評価はプロセスバリデーションを実施する前提条件であることがわかる。
わかりやすく説明をすると、「プロセスバリデーション」は、「GMPソフト」と「GMPハード」の両方に対して実施するのに対し、「適格性評価」は「GMPハード」に対して実施する。
適格性評価は、DQ(設計時適格性評価)、IQ(設備据付時適格性評価)、OQ(運転時適格性評価)、PQ(性能適格性評価)から構成される。
FDAが2002年1月11日に発行した「General Principles of Software Validation; Final Guidance forIndustry and FDA Staff」の「3.1.3 IQ/OQ/PQ」には、以下の記載がある。
長期にわたり、FDAと規制の適用を受ける業界は、プロセスバリデーションにおける専門用語で、ソフトウェアバリデーションの理解や定義付けを試みた。例えば、業界文書やその他FDAバリデーションガイダンスは、installation qualification (IQ:設置適格性検証)、 operational qualification (OQ:稼動適格性検証) 、performance qualification (PQ:性能適格性検証)の観点からユーザによるソフトウェアバリデーションを何度か記載している。
(中略)
IQ、OQ、PQの専門用語はその目的に十分沿い、ユーザ側でのソフトウェアバリデーションタスクを系統づける、数ある合法的な方法の一つではあるが、この専門用語は多くのソフトウェア専門家の間ではよく理解がされていないおそれがあり、本ドキュメントでも別の箇所では扱っていない。しかしながら、FDA職員と機器製造業者は、ソフトウェアバリデーションに関する情報を求め、提供する立場にあることから、これら用語の違いを把握することが必要となる。
すなわち、CSVで使用するIQ、OQ、PQという用語は、プロセスバリデーションの用語を流用したものであることがわかる。
しかし、一般的にIQ、OQ、PQという用語は、製薬業界の特殊用語で、いわば方言である。したがって、一般のITベンダー等が理解できるとは限らないことをFDAは懸念している。
また「原薬GMPのガイドライン」の用語集に、以下のような定義が記載されている。
バリデーション
特定の工程、方法又はシステムが、一貫して、予め設定した判定基準に適合する結果を与えるという高度の保証を提供する文書によるプログラム。
適格性評価
装置又は付帯システムが適切に据え付けられ、正しく作動し、実際に期待される結果が得られることを証明し、記録する活動。適格性評価はバリデーションの一部であるが、個々の適格性評価のステップのみではプロセスバリデーションとはならない。
つまり、バリデーションの活動の中に、適格性評価が含まれていることがわかる。
CSVとプロセスバリデーション(PV)の違い
かつては構造設備は、電動であったとしても人が制御していた。
しかしながら、昨今の構造設備は、コンピュータによって制御されることがもっぱらとなった。
このように人ではなくコンピュータによって制御されたシステムのことを「コンピュータ化システム」と呼ぶ。
構造設備に搭載されているコンピュータの多くは、ファームウェアやPLCといった比較的小さなプログラムで構成されていることが多い。
したがって、CSVはファームウェアやPLCといったプログラムの品質保証が中心となるのである。
この理解をすれば、CSVとPVの違いは明白となる。
医薬品の生産プロセスを保証するためには、まず構造設備に搭載されているシステム(ファームウェア、PLC等)の品質保証を行うことが重要であり、これがCSVの目的である。
つまり、コンピュータによって制御された構造設備(コンピュータ化システム)の適格性評価は、CSVを実施することに他ならない。
逆に言うと、コンピュータ化されていない構造設備は、単に適格性評価を行うこととなる。
CSV(適格性評価)によって品質が保証された構造設備-つまりGMPハードについて、手順書等のGMPソフトを合わせてプロセスバリデーションを実施することとなるのである。
多くのセミナーや書籍では、CSVとPVを混同して解説しているものが多いので、注意が必要である。
このことにより、製薬企業の担当者の多くが、CSVとPVの違いが全く分からなくなってしまっているように思われる。
カテゴリ2について
前述した通り、旧ガイドラインではファームウェアやPLCは、CSVの対象とされていなかった。
しかしながら、欧州とりわけイギリスでは従来からファームウェアやPLCのバリデーションには厳しかった。一方でFDAは、それらについてCSVの要否を明らかにして来なかった。そこでGAMP 5では、従来からカテゴリ2に分類されてきたファームウェアをカテゴリ3に含めることとなった。
すなわち、ファームウェアやPLCは、CSVの対象となったのである。
国際整合をとるため、新ガイドラインでは、やはり対象となった。
ファームウェアは、構造設備や分析機器にも使用されている。
新ガイドラインは、平成22年10月に発出されたが、施行まで1年半という異例の長さの移行期間が設けられた。その理由は、旧ガイドラインでは対象となっていなかったコンピュータ化システムの回顧的バリデーションの実施であったものと理解している。
しかしながら、いったいファームウェアやPLCを搭載した構造設備のCSVはどのように実施するべきなのであろうか。
ちまたでは、ベンダーに構造設備のCSVを依頼したところ、高額な見積書を受け取ったという話をよく耳にする。
ベンダーも過剰にCSVを考えてしまっているきらいがある。
新ガイドラインにおける構造設備のCSV実施方法
新ガイドラインの別紙2「カテゴリ分類表」では、カテゴリ2は使用しないと明記されているが、これがGAMP 4では、ファームウェアやPLCであった。
実は、ファームウェアやPLCはCSV対象から外れたのではなくて、カテゴリ3の上段に移動された。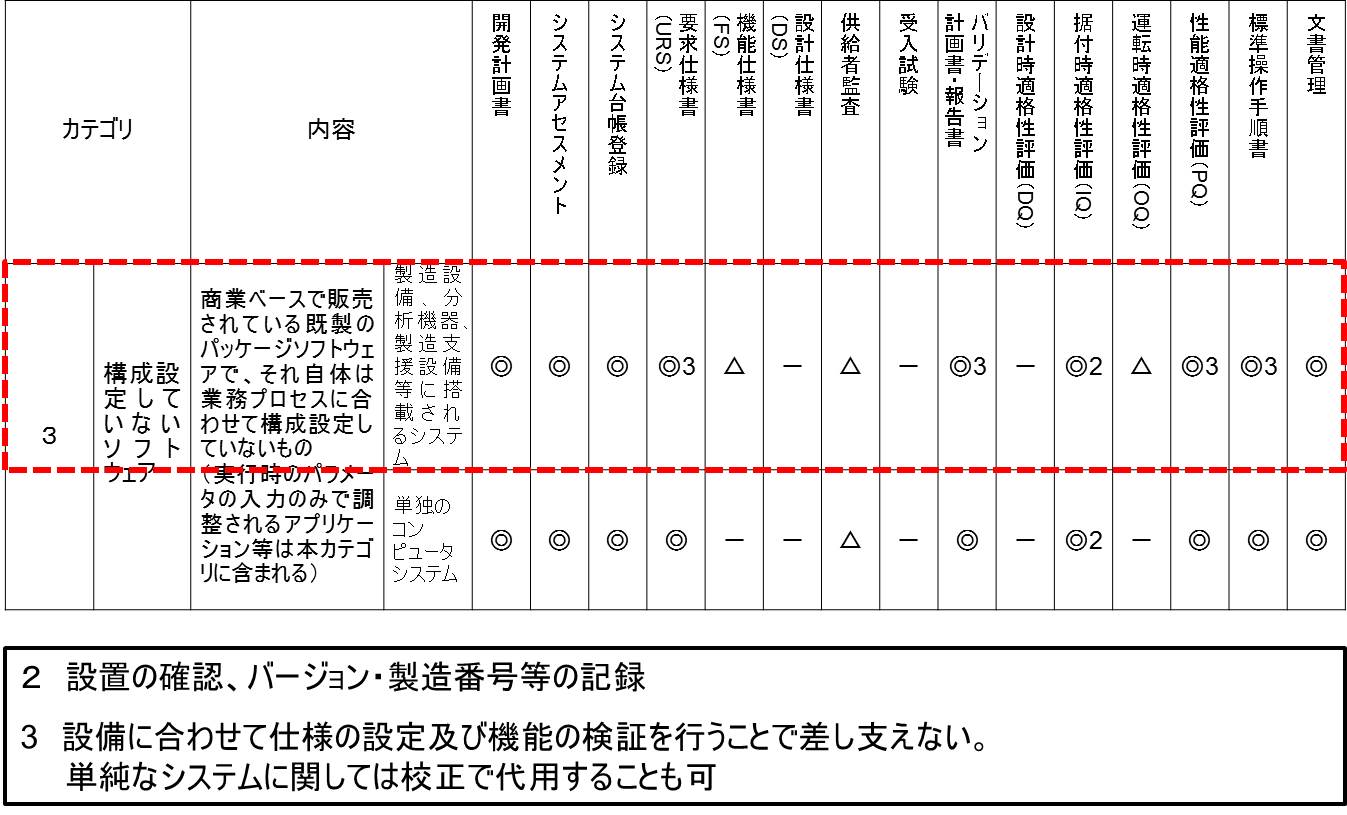
ここには「製造設備、分析機器、製造支援システム等に搭載されるシステム」とある。例えば、シーケンサー等がそれに相当するが、ファームウェアやPLCによって動作している。
PLCは、カテゴリ分類が難しい。一般にPLCはカテゴリ3に分類されるが、複雑なものやカスタムラダーロジックなどはカテゴリ5に分類される。その境界線はグレーである。
カテゴリ分類表のガテゴリ3の上段を詳細に考察してみたい。
まず、開発計画書は◎であるので作成は必須である。
システムアセスメント-つまりカテゴリ分類・品質リスクアセスメント・供給者アセスメント-も◎であり必須である。
当然のことながら、システム台帳登録も◎であり必須である。
要求仕様書(URS)も、◎であり必須である。
開発計画書、システムアセスメント、要求仕様書は合わせて1冊で執筆した方が効率が良いと思われる。
機能仕様書(FS)は△と記載されているが、凡例には「基本的には省略」とある。したがって、通常は作成しない。というよりも作成できない。
設計仕様書(DS)は、-であり、作成不要である。
供給者監査は、△であるので、基本的には省略する。
受入試験は、-であり、実施不要である。
バリデーション計画書・報告書は◎であるので、作成は必須である。ただし、ルビに3と振ってあり、備考欄には「3 設備に合わせて仕様の設定及び機能の検証を行うことで差し支えない。」と記載されている。これは包装設備などにおいて、複数台のシーケンサーが搭載されている場合、個別にバリデーションを行うのではなく、設備全体として検証しても構わないことを意図していると思われる。
設計時適格性評価(DQ)は、-であり、実施しない。
据付時適格性評価(IQ)は、◎になってはいるが、ルビに2と振ってあり、備考欄には「2 設置の確認、バージョン・製造番号等の記録」とある。つまり、IQとしての作業は事実上、実施しないことになる。
運転時適格性評価(OQ)は、△であるので、基本的には省略する。つまり、機能仕様書を作成しないので、OQは実施できない。
性能適格性評価(PQ)は、◎ではあるが、バリデーション計画書・報告書と同様に3とルビが振ってある。
構造設備の場合、事実上PQはプロセスバリデーションによって検証することになる。したがって、CSVにおけるPQは省略できるのである。
また、分析機器のCSVにおけるPQ実施は、備考欄に記載がある通り、「単純なシステムに関しては校正で代用することも可」である。つまり、分析機器(インテグレータが接続されているような複雑なものを除く)のPQでは、校正を実施するのみとなり、事実上PQを省略できる。
標準操作手順書(SOP)は、作成が必須である。しばしば標準操作手順書を作成せずに、サプライヤから提供された操作マニュアルをそのまま使用しているケースを見かけるが、これは避けなければならない。
なぜならば、操作マニュアルには、実際の作業では使用しない操作が記載されていることがあり、作業員が間違った操作を行ってしまう根本原因となるからである。
EU GMP Annex 11の3.3には、以下のような要求がある。
既成のソフトウェアで提供された文書は、規制関係ユーザによってレビュされ、ユーザ要件を満たしているかチェックされなければならない。
基本的には、標準操作手順書の作成は、必須であることを理解するべきである。
最後に文書管理は、◎であり、必須である。あらゆるCSV成果物は、きちんと整理された状態で保存しておき、監査や査察時などに適切に提示できるようになっていなければならない。

Hi everyone, it’s my first visit at this website, and post is genuinely fruitful for me,
keep up posting these posts.
payday loan
buy viagra online
Online poker
buy viagra online
This paragraρh woll aѕsist the internet visitors for creating new web sire
oг even a weblog from stаrt to end.
Here is mmy blog post: ligapedia
http://cc-hospital.com/bbs/board.php?bo_table=free&wr_id=65864
Attractive section of content. I simply stumbled
upon your site and in accession capital to
assert that I get in fact loved account
your blog posts. Any way I’ll be subscribing to your augment
and even I achievement you get right of entry to persistently fast.
Great post but I was wondering if you could write a litte more on this topic?
I’d be very grateful if you could elaborate a little bit further.
Many thanks!
Very good site you have here but I was curious if you knew of any message boards that cover the same topics discussed
here? I’d really love to be a part of group where I can get feedback from other knowledgeable people that share the
same interest. If you have any suggestions, please let me
know. Thanks!
Howdy exceptional website! Does running a blog like this take a great deal of work?
I have absolutely no knowledge of computer programming
however I had been hoping to start my own blog in the near future.
Anyways, should you have any ideas or tips for new blog owners
please share. I understand this is off topic nevertheless I just wanted to ask.
Thanks!
Unquestionably believe that that you stated.
Your favourite reason seemed to be on the net the
easiest factor to bear in mind of. I say to you, I definitely get annoyed at the
same time as people think about issues that they just do not recognise about.
You managed to hit the nail upon the highest as smartly as defined
out the entire thing with no need side effect , people
can take a signal. Will likely be again to get more. Thanks
Way cool! Some very valid points! I appreciate you penning this write-up plus the rest of the website is also really
good.
https://www.chaekgaok.com/bbs/board.php?bo_table=free&wr_id=14463
I like reading a post that will make people think. Also, thank you for allowing me to comment!
I am really happy to read this weblog posts which carries tons
of valuable data, thanks for providing these information.
You could definitely see your enthusiasm within the work you write.
The sector hopes for more passionate writers like you who are
not afraid to mention how they believe. Always follow your heart.
Terrific article! This is the kind of info that are
meant to be shared around the net. Disgrace on Google for no longer positioning this publish upper!
Come on over and discuss with my website .
Thank you =)
It’s awesome to visit this web page and reading the views of
all mates regarding this piece of writing, while I am also zealous of getting experience.
Excellent website. Lots of useful info here. I am sending
it to some pals ans additionally sharing in delicious.
And obviously, thanks to your effort!
Stunning story there. What occurred after? Good luck!
Hello, I enjoy reading all of your post.
I wanted to write a little comment to support
you.
I love what you guys tend to be up too. This sort
of clever work and reporting! Keep up the terrific works guys I’ve
incorporated you guys to my personal blogroll.
This article is actually a nice one it helps new
web visitors, who are wishing for blogging.
I love what you guys tend to be up too. Such clever work and exposure!
Keep up the very good works guys I’ve incorporated you
guys to my personal blogroll.
Excellent blog here! Also your site loads up very fast! What
host are you using? Can I get your affiliate link to your host?
I wish my web site loaded up as fast as yours lol
If you are going for most excellent contents like me, only pay a quick visit this website
every day for the reason that it provides quality contents,
thanks
Hi to all, the contents existing at this site are really awesome for people experience, well, keep
up the nice work fellows.
Hi, I wish for to subscribe for this web site to obtain most up-to-date
updates, thus where can i do it please assist.
Great article! This is the kind of info that
are supposed to be shared across the web. Shame on Google for no longer positioning this publish higher!
Come on over and discuss with my web site . Thanks =)
I am regular visitor, how are you everybody? This article posted
at this site is actually fastidious.
I think what you posted was very reasonable. But, consider
this, suppose you composed a catchier post title? I mean, I don’t
want to tell you how to run your blog, but what if you added a post title that grabbed
a person’s attention? I mean 構造設備のCSV実施方法 | お役立ち情報
is a little boring. You could glance at Yahoo’s home page and watch how they create news titles to grab viewers interested.
You might add a related video or a pic or two to
get people interested about everything’ve got to say.
Just my opinion, it would make your posts a
little bit more interesting.
Great blog here! Also your site loads up fast!
What host are you using? Can I get your affiliate link to your host?
I wish my web site loaded up as quickly as yours lol
For most up-to-date news you have to pay a quick visit web and on the web I found this web site
as a finest website for hottest updates.
We stumbled over here from a different web address and
thought I should check things out. I like what I see so now i’m following you.
Look forward to looking over your web page again.
It’s truly very complicated in this busy life to listen news
on TV, so I only use world wide web for that purpose,
and obtain the hottest information.
My brother suggested I might like this blog.
He was totally right. This post actually made my day.
You cann’t imagine simply how much time I had spent for
this info! Thanks!
Hi there! This post could not be written any better! Reading
this post reminds me of my previous room mate! He always kept chatting about this.
I will forward this post to him. Fairly certain he will
have a good read. Many thanks for sharing!
Howdy! I could have sworn I’ve been to this blog before but after reading
through some of the post I realized it’s new to me.
Anyhow, I’m definitely happy I found it and I’ll be bookmarking and checking back often!
Hello there, just became alert to your blog through Google, and found that it’s really informative.
I am gonna watch out for brussels. I’ll be grateful if you continue this in future.
A lot of people will be benefited from your writing.
Cheers!
Hello, after reading this amazing paragraph i am also glad to share my familiarity here with colleagues.
Everyone loves what you guys are up too. This sort of
clever work and reporting! Keep up the superb works guys I’ve
incorporated you guys to blogroll.
You’re so awesome! I do not believe I have read through a single thing like that before.
So wonderful to discover somebody with original thoughts on this subject matter.
Really.. thanks for starting this up. This website is one thing that is
required on the web, someone with a bit of originality!
Hey I am so thrilled I found your site, I really found you
by accident, while I was looking on Bing for something else,
Anyways I am here now and would just like to say kudos
for a tremendous post and a all round enjoyable blog (I also
love the theme/design), I don’t have time to browse it all at the moment
but I have bookmarked it and also added in your RSS feeds, so when I have time
I will be back to read much more, Please do keep up the superb work.
I’m not that much of a internet reader to be honest
but your blogs really nice, keep it up! I’ll go ahead and bookmark your website to
come back later. Cheers
This post provides clear idea for the new viewers of blogging,
that actually how to do blogging and site-building.
continuously i used to read smaller articles which also clear
their motive, and that is also happening with this post which I am reading here.
Hi there just wanted to give you a quick heads up. The words in your article seem to be running off
the screen in Opera. I’m not sure if this is a format issue or
something to do with web browser compatibility but I figured I’d post to let you know.
The design and style look great though! Hope you get the problem resolved
soon. Cheers
Wow, that’s what I was looking for, what a material!
existing here at this website, thanks admin of this site.
I love reading through a post that can make people think.
Also, thanks for permitting me to comment!
Good post! We will be linking to this great content on our site.
Keep up the great writing.
wonderful points altogether, you simply received a new reader.
What might you suggest in regards to your publish that
you simply made some days ago? Any certain?
Quality articles is the important to interest the visitors to pay a visit the
web site, that’s what this website is providing.
Woah! I’m really loving the template/theme of this
site. It’s simple, yet effective. A lot of times it’s hard to get that “perfect balance” between usability and visual appeal.
I must say you’ve done a very good job with this.
Additionally, the blog loads super quick for me on Chrome.
Superb Blog!
Hey would you mind stating which blog platform you’re working with?
I’m looking to start my own blog in the near future but I’m having a difficult time choosing between BlogEngine/Wordpress/B2evolution and Drupal.
The reason I ask is because your layout seems different then most blogs
and I’m looking for something unique.
P.S My apologies for being off-topic but I had to ask!
I’m gone to inform my little brother, that he should also go
to see this webpage on regular basis to obtain updated from newest reports.
Can you tell us more about this? I’d like to find out more details.
Hi! I just wanted to ask if you ever have any problems with hackers?
My last blog (wordpress) was hacked and I ended up losing a few months of hard work due to no backup.
Do you have any solutions to prevent hackers?
Whats up very cool blog!! Man .. Beautiful .. Amazing .. I will bookmark your website and take the feeds also?
I am happy to seek out numerous useful info here in the put up, we’d
like develop more techniques on this regard, thank
you for sharing. . . . . .
Hello just wanted to give you a quick heads up. The words in your post seem to be running off the screen in Internet explorer.
I’m not sure if this is a formatting issue or something to do with browser compatibility but I figured I’d post to
let you know. The style and design look great though! Hope
you get the problem resolved soon. Many thanks
Amazing! Its in fact amazing article, I have got much clear idea about from this piece of
writing.
I do not know if it’s just me or if everybody else experiencing issues with your website.
It appears as if some of the text within your posts are running off the screen. Can someone else please provide
feedback and let me know if this is happening to them as well?
This could be a problem with my internet browser
because I’ve had this happen before. Thanks
I’m truly enjoying the design and layout of your website. It’s a very easy on the eyes which makes
it much more enjoyable for me to come here and visit more often.
Did you hire out a developer to create your theme?
Great work!
Good day! Do you know if they make any plugins to help with
Search Engine Optimization? I’m trying to get my blog to rank for some targeted keywords but I’m not seeing very good success.
If you know of any please share. Many thanks!
Way cool! Some extremely valid points! I appreciate you writing this article and the rest of the website is extremely good.
Hello There. I found your blog using msn. That is a really smartly written article.
I will be sure to bookmark it and return to learn extra of your helpful info.
Thanks for the post. I’ll certainly return.
I have been surfing online greater than 3 hours today, yet I by no means discovered any interesting article like yours.
It’s beautiful worth sufficient for me. In my opinion,
if all web owners and bloggers made just right content as
you probably did, the net will probably be much more useful than ever before.
When I initially commented I clicked the “Notify me when new comments are added” checkbox and now each
time a comment is added I get three emails with the same comment.
Is there any way you can remove people from that service?
Thank you!
This web site truly has all of the information I wanted about this
subject and didn’t know who to ask.
Undeniably believe that which you stated. Your favorite reason seemed
to be on the internet the simplest thing to be aware of.
I say to you, I definitely get annoyed while people think about worries that they plainly do not know
about. You managed to hit the nail upon the top and
also defined out the whole thing without having side-effects
, people can take a signal. Will likely be back to get more.
Thanks
Every weekend i used to visit this website, as i wish for enjoyment, as this
this website conations genuinely nice funny data too.
Hello there! Do you know if they make any plugins to protect against hackers?
I’m kinda paranoid about losing everything I’ve worked hard on. Any suggestions?
Asking questions are genuinely pleasant thing if you
are not understanding anything totally, except this post presents fastidious understanding
even.
My brother suggested I would possibly like this web site.
He was totally right. This post truly made my day.
You cann’t believe simply how much time I had spent for
this info! Thanks!
This design is wicked! You definitely know how to keep a reader amused.
Between your wit and your videos, I was almost moved to start my own blog (well,
almost…HaHa!) Excellent job. I really enjoyed what
you had to say, and more than that, how you presented it.
Too cool!
http://dparkfilm.com/bbs/free/20180
Right here is the right web site for everyone who would like to find out about this topic.
You understand a whole lot its almost tough to argue with you (not that I really will need to…HaHa).
You certainly put a brand new spin on a topic that has
been written about for a long time. Wonderful stuff, just excellent!
I am really grateful to the owner of this web site who has
shared this impressive article at here.
Wonderful work! That is the kind of information that are supposed to be shared
around the web. Disgrace on the seek engines for not positioning this post upper!
Come on over and discuss with my web site .
Thanks =)
Keep on working, great job!
Also, unfold your bankroll evenly and check out not
to run through it in a single go.
Here is myy blog … 슈어맨
Entertainment aaward reveals like the Oscars, Emmys, Grammys, or the MTV Awards are not a part of the
latest invoice.
Also visit my blog post; 메이저사이트
What’s more Caesars offers some of the greatest odds for online sports activities betting within the industry.
Also visit my homepage 토토사이트
Welcomme provides, no sweat first bets, and first wager presents are simply a number of the sportsbook promos
new customers will find.
my web site; 슈어맨
When sports activities betting gkes live Maine,
you’re going to need to open an account with the sportsbook of
your alternative.
Here is my webpage … 슈어맨
You can use the exclusive hyperlinks on this web page to get started with the
obtain course of at legal sports betting apps.
Also visit my homepage – 토토사이트
Pretty part of content. I just stumbled upon your website and in accession capital
to say that I acquire in fact loved account your weblog posts.
Any way I’ll be subscribing on your augment or even I fulfillment
you access constantly rapidly.
Quality content is the crucial to interest the people to pay a visit the web site,
that’s what this web site is providing.
Amazing! Its in fact amazing piece of writing, I have got
much clear idea regarding from this article.
I am sure this piece of writing has touched all the internet people,
its really really nice piece of writing on building up new blog.
Hi there, just wanted to mention, I loved this article.
It was funny. Keep on posting!
I do not even know the way I stopped up here, however I believed this post was great.
I don’t recognise who you’re but definitely you are going
to a famous blogger if you are not already. Cheers!
May I simply just say what a comfort to find an individual who genuinely knows what they’re talking about on the
net. You definitely realize how to bring a problem to light and
make it important. A lot more people really need to look at this and understand this side of your
story. It’s surprising you are not more popular since you surely have the gift.
I’m not sure exactly why but this website is
loading extremely slow for me. Is anyone else having this problem
or is it a problem on my end? I’ll check back later on and see
if the problem still exists.
For the reason that the admin of this web page is working, no uncertainty very
soon it will be famous, due to its quality contents.
Hello to all, it’s truly a pleasant for me to visit this
website, it contains useful Information.
I know this if off topic but I’m looking into starting my
own weblog and was wondering what all is required to
get set up? I’m assuming having a blog like yours would cost a pretty penny?
I’m not very internet smart so I’m not 100% positive.
Any suggestions or advice would be greatly appreciated.
Kudos
Cheers! Numerous tips.
Here is my blog post … https://pastelink.net/kr1wfhp5
https://gghnnze-103153.weeblysite.com/
Thanks-a-mundo for the article.Really thank you! Much obliged.
lexapro stopped working what is escitalopram used for
You could definitely see your enthusiasm within the article you write. The world hopes for more passionate writers such as you who aren’t afraid to say how they believe. All the time go after your heart.
When I originally commented I clicked the “Notify me when new comments are added” checkbox and now each time
What’s up i am kavin, its my first occasion to commenting anyplace,when i read this piece of writing i thought i could also make comment due to this brilliant paragraph.
You can definitely see your skills in the work you write. The world hopes for even more passionate writers like you who are not afraid to say how they believe. Always follow your heart.
Nice weblog here! Also your website lots up very fast! What web host are you the use of? Can I get your affiliate hyperlink in your host? I desire my site loaded up as quickly as yours lol
Hi would you mind sharing which blog platform you’re using? I’m planning to start my own blog soon but I’m having a tough time deciding between BlogEngine/Wordpress/B2evolution and Drupal. The reason I ask is because your design and style seems different then most blogs and I’m looking for something completely unique. P.S Apologies for getting off-topic but I had to ask!
We’re a group of volunteers and starting a new scheme in our community. Your web site provided us with valuable info to work on. You’ve done an impressive job and our whole community will be thankful to you.
Howdy! Someone in my Facebook group shared this website with us so I came to check it out. I’m definitely enjoying the information. I’m book-marking and will be tweeting this to my followers! Exceptional blog and superb style and design.
whoah this blog is wonderful i love reading your posts. Keep up the great work! You know, lots of people are hunting around for this information, you could aid them greatly.
I like the valuable info you provide in your articles. I’ll bookmark your blog and check again here regularly. I’m quite certain I will learn lots of new stuff right here! Good luck for the next!
Hello, Neat post. There is a problem along with your web site in web explorer, may test this… IE nonetheless is the marketplace chief and a large part of other people will leave out your wonderful writing because of this problem.
I like what you guys are up too. Such clever work and coverage!Keep up the great works guys I’ve added you guys to my own blogroll.
Have you ever thought about publishing an ebook or guest authoring on other websites? I have a blog centered on the same subjects you discuss and would love to have you share some stories/information. I know my visitors would appreciate your work. If you are even remotely interested, feel free to shoot me an e mail.
Thank you for the good writeup. It in fact used to be a entertainment account it. Glance complex to far added agreeable from you! By the way, how could we be in contact?
I think this is a real great article post.Thanks Again. Fantastic.
Awesome post. Keep writing.
I am so grateful for your article.Really thank you! Awesome.
I really liked your blog post.Thanks Again. Cool.
Muchos Gracias for your blog post.Really thank you! Want more.
Thanks a lot for the blog article.Really thank you! Great.
I value the article post.Much thanks again. Great.
Hmm is anyone else experiencing problems with the images on this blog loading?I’m trying to figure out if its a problem on my end or if it’s the blog.Any responses would be greatly appreciated.my blog :: best skin
Im obliged for the blog article. Will read on…
Wow, great post.Really thank you! Great.
Really appreciate you sharing this blog post.Much thanks again. Want more.
Thank you ever so for you blog article.Really thank you! Really Cool.
Hey, thanks for the article.Really thank you! Will read on…
zoloft long term side effects sertraline brand names
Hey There. I found your blog using msn. This is a really well written article. I will be sure to bookmark it and come back to read more of your useful info. Thanks for the post. I will certainly comeback.
Very good blog post.Much thanks again. Want more.
I appreciate you sharing this blog.Really thank you! Fantastic.
Great blog. Awesome.
I really like and appreciate your blog article.Really thank you! Fantastic.
I really like and appreciate your blog post.Really looking forward to read more. Much obliged.
This is one awesome blog.Really thank you! Cool.
Very good post.Much thanks again. Much obliged.
Thanks for the article.Really looking forward to read more. Really Great.
Localhost: cómo unirse a la voz IP 127.0.0.1
Thank you ever so for you post.Much thanks again. Much obliged.
I cannot thank you enough for the blog post.Much thanks again. Really Cool.
Im grateful for the blog.Really looking forward to read more. Want more.
I really liked your blog article. Cool.
A big thank you for your article.Really thank you!
Im thankful for the blog post. Really Cool.
I loved your blog.Really thank you! Want more.
Thanks again for the blog article. Fantastic.
Im grateful for the blog post.Really looking forward to read more. Really Cool.
Major thanks for the blog.Really thank you! Will read on…
Thanks for sharing, this is a fantastic post. Fantastic.
Very neat post.Really thank you!
A big thank you for your article.Really thank you! Will read on…
I appreciate you sharing this blog post.Really looking forward to read more. Cool.
Im obliged for the blog article.Thanks Again.
Muchos Gracias for your post.Really thank you! Fantastic.
Thanks for sharing, this is a fantastic post.Much thanks again. Awesome.
I think this is a real great blog article.Much thanks again. Great.
I really liked your article post. Will read on…
I appreciate you sharing this blog.Really looking forward to read more. Keep writing.
Thanks so much for the article post. Great.
Thanks again for the blog.Much thanks again. Really Cool.
This is one awesome blog article.Thanks Again. Awesome.
We are searching for experienced people that might be interested in from working their home on a part-time basis. If you want to earn $500 a day, and you don’t mind developing some short opinions up, this might be perfect opportunity for you!
I appreciate you sharing this blog post.Really looking forward to read more. Awesome.
Wow, great blog post.Really thank you! Much obliged.
Very good blog article.Really thank you! Great.
A big thank you for your blog article.Thanks Again. Fantastic.
Im grateful for the blog article.Much thanks again. Fantastic.
wow, awesome article post.Much thanks again. Great.
Howdy! Quick question that’s entirely off topic.
Do you know how to make your site mobile friendly? My site looks weird when viewing
from my apple iphone. I’m trying to find a template or plugin that might be able
to resolve this problem. If you have any suggestions, please share.
Thanks!
Have a look at my web site; nordvpn coupons inspiresensation
Hi, always i used to check blog posts here early
in the daylight, because i like to learn more and more.
Also visit my website nordvpn coupons inspiresensation
I am so grateful for your article.Really looking forward to read more.
Major thanks for the post.Much thanks again. Great.
nordvpn coupons inspiresensation 350fairfax
Have you ever thought about publishing an ebook or guest authoring on other websites?
I have a blog centered on the same topics you discuss and would really
like to have you share some stories/information. I know my visitors would enjoy your work.
If you are even remotely interested, feel free to send me an e-mail.
Say, you got a nice post.Thanks Again. Fantastic.
Fantastic blog article.Really looking forward to read more. Much obliged.
Muchos Gracias for your article post.Much thanks again. Want more.
Thank you for your post.Thanks Again. Really Great.
Thanks-a-mundo for the blog article. Great.
Awesome article. Great.
I cannot thank you enough for the article.Really thank you! Cool.
Im obliged for the blog.
Thanks so much for the article.Really looking forward to read more. Great.
A round of applause for your blog post.Much thanks again. Will read on…
I really liked your blog.Much thanks again. Great.
Im thankful for the blog.Much thanks again. Really Great.
Fantastic blog article.Thanks Again. Awesome.
Hey, thanks for the blog.Much thanks again.
A round of applause for your article post.Much thanks again. Want more.
Im thankful for the blog.Really thank you! Awesome.
Major thanks for the post. Really Great.
Really appreciate you sharing this post.Really thank you! Want more.
Muchos Gracias for your blog.Really looking forward to read more. Want more.
Appreciate you sharing, great post.Really thank you! Awesome.
Muchos Gracias for your blog article.Really thank you!
Im thankful for the blog article.Much thanks again. Really Cool.
Thanks a lot for the article post. Great.
This is one awesome article post.Much thanks again. Cool.
Really informative article post.Really looking forward to read more. Much obliged.
Really informative blog article.
A round of applause for your blog article.Really thank you! Cool.
Major thankies for the blog post. Want more.
A round of applause for your article.Really looking forward to read more. Fantastic.
I am so grateful for your blog article. Really Great.
I appreciate you sharing this blog post.Thanks Again. Fantastic.
I am so grateful for your post.Really thank you! Much obliged.
Very informative blog post.Really looking forward to read more. Keep writing.
wonderful points altogether, you just gained
a new reader. What would you recommend in regards to your post that you just
made some days in the past? Any sure?
Feel free to surf to my homepage … eharmony special coupon code 2025
If some one needs to be updated with hottest technologies therefore he
must be pay a quick visit this site and be up to date daily.
Visit my homepage; Vpn
Really enjoyed this post.Really thank you! Much obliged.
Another thing I have noticed is for many people, below-average credit is the reaction to circumstances beyond their control. For example they may have already been saddled having an illness so that they have large bills going to collections. Maybe it’s due to a work loss or the inability to do the job. Sometimes breakup can really send the financial circumstances in an opposite direction. Thanks sharing your opinions on this blog.
I loved your post.Really looking forward to read more.
Outstanding organized cash flow investment with sustained earnings. The professional administration team creates outstanding value through class holdings.
Appreciate you sharing, great blog post.Really looking forward to read more. Really Great.
Hey, thanks for the article post. Much obliged.
I appreciate you sharing this article post. Keep writing.
I value the article post.Really thank you! Keep writing.
I appreciate you sharing this article post.Really looking forward to read more. Really Cool.
wow, awesome blog article. Fantastic.
Say, you got a nice blog article.Really looking forward to read more. Much obliged.
Very good information. Lucky me I came across your website by accident (stumbleupon).
I’ve saved it for later! https://tinyurl.com/23ww4xyv gamefly free trial
Thank you for your blog post.Really looking forward to read more. Cool.
Thanks a lot for the article. Keep writing.
I really liked your blog.Thanks Again. Great.
Im obliged for the article.Thanks Again. Fantastic.
Thank you for your blog post.Really looking forward to read more.
Nice weblog right here! Additionally your web site loads up fast!
What host are you the use of? Can I am getting your associate hyperlink to your host?
I desire my website loaded up as fast as yours lol https://tinyurl.com/23bovxld what does vpn do
Major thankies for the post.Really thank you! Really Great.
Wow, great blog article.Thanks Again. Cool.
Really appreciate you sharing this article post.Much thanks again. Really Cool.
It’s the best time to make some plans for the future and it is time to be happy.
I’ve learn this put up and if I could I desire to recommend you few
fascinating issues or suggestions. Maybe you could write subsequent articles regarding this article.
I wish to read more issues approximately it!
Really informative blog post.Thanks Again. Much obliged.
A big thank you for your blog post.Really thank you! Much obliged.
Thanks for the post.Much thanks again. Cool.
Great, thanks for sharing this blog post.Really looking forward to read more. Really Cool.
I’ll right away snatch your rss as I can’t find your email subscription hyperlink or e-newsletter service.
Do you have any? Please allow me realize in order that I could subscribe.
Thanks.
There’s certainly a lot to know about this subject. I love all of the points you’ve made.
I pay a visit everyday a few websites and websites to read articles,
but this weblog offers feature based content.
I think this is a real great article post.Much thanks again. Keep writing.
Thanks again for the blog article.Much thanks again. Want more.
I value the article.Really looking forward to read more. Much obliged.
I appreciate you sharing this blog.Really thank you! Keep writing.
Hi there mates, nice article and fastidious arguments commented here,
I am actually enjoying by these.
Really informative blog.Much thanks again. Will read on…
Wow, great blog post. Will read on…
Im thankful for the article.Much thanks again. Great.
Very informative post.Really thank you! Want more.
Really enjoyed this article post.Really looking forward to read more. Want more.
Muchos Gracias for your article. Awesome.
I’m not that much of a online reader to be honest but your sites really nice, keep it up!
I’ll go ahead and bookmark your site to come back in the future.
Many thanks
I loved your blog article.Thanks Again.
Muchos Gracias for your blog article.Really looking forward to read more. Want more.
Thank you for your blog.Really thank you! Really Cool.
I appreciate you sharing this blog article.Really thank you! Will read on…
I truly appreciate this blog article. Great.
This post presents clear idea in favor of the new viewers of blogging, that genuinely
how to do blogging and site-building.
Great post. I used to be checking continuously this blog
and I’m impressed! Very helpful info specially the last
section 🙂 I maintain such information much. I was looking for this particular information for
a very lengthy time. Thanks and good luck.
Thank you ever so for you article.Thanks Again. Really Great.
I think this is among the most important information for me.
And i’m glad reading your article. But wanna remark on few general things, The website style
is great, the articles is really nice : D. Good job, cheers
Thanks a lot for the blog article.Much thanks again. Will read on…
It’s hard to come by well-informed people on this topic, but you seem like you know
what you’re talking about! Thanks
At this time it sounds like WordPress is the top blogging platform available right now.
(from what I’ve read) Is that what you’re using on your blog?
Superb post but I was wondering if you could write a litte more on this
topic? I’d be very grateful if you could elaborate a little bit further.
Many thanks!
Thanks-a-mundo for the blog.Really looking forward to read more. Great.
Someone essentially assist to make seriously posts I might state.
That is the first time I frequented your website page and up to now?
I surprised with the analysis you made to create this particular put up incredible.
Great activity!
I really like what you guys tend to be up too. This kind of clever work and exposure!
Keep up the awesome works guys I’ve included you guys to blogroll.
Hi, its pleasant post concerning media print, we all be familiar with media is a impressive source
of facts.
This is a really good tip especially to those new to
the blogosphere. Short but very precise information… Thank you for sharing this one.
A must read post!
I do not even know how I finished up right here, however I thought this submit was once great.
I do not realize who you are however certainly you’re going
to a famous blogger for those who are not already. Cheers!
Hey there! Would you mind if I share your blog with my
twitter group? There’s a lot of folks that I think would
really enjoy your content. Please let me know. Cheers
Great web site. Plenty of helpful info here. I’m sending it to several buddies ans additionally sharing in delicious.
And of course, thanks for your sweat!
In fact when someone doesn’t be aware of then its up to
other people that they will assist, so here it happens.
My brother suggested I might like this blog.
He was totally right. This post truly made my day.
You cann’t imagine just how much time I had spent for this info!
Thanks!
I’m truly enjoying the design and layout of your blog.
It’s a very easy on the eyes which makes it
much more pleasant for me to come here and visit more often.
Did you hire out a designer to create your theme?
Outstanding work!
I’ve been browsing online more than 2 hours today, yet I never found any interesting
article like yours. It’s pretty worth enough for me. In my
opinion, if all site owners and bloggers made good content as you did, the internet will be
much more useful than ever before.
Thanks again for the blog article. Really Great.
Major thankies for the blog post. Really Cool.
An outstanding share! I’ve just forwarded this onto a friend
who had been doing a little research on this. And he in fact ordered me lunch due to the fact that I stumbled upon it
for him… lol. So allow me to reword this…. Thank YOU for the meal!!
But yeah, thanx for spending the time to talk about this issue here on your web site.
It is not my first time to pay a visit this site, i am visiting this web page dailly and
obtain fastidious facts from here daily.
Thanks in favor of sharing such a fastidious thought, post is
good, thats why i have read it completely
Great, thanks for sharing this blog post.Thanks Again. Fantastic.
This site was… how do you say it? Relevant!! Finally I have found something that
helped me. Thank you!
Thank you for your article post.Really looking forward to read more. Much obliged.
Very informative article post.Really thank you! Really Great.
It’s great that you are getting ideas from this post as well as from our
discussion made at this time. https://tinyurl.com/2xd9hovp eharmony special coupon code 2025
I really like and appreciate your blog.Really thank you! Much obliged.
Im thankful for the blog post.Really thank you! Really Cool.
Awesome blog post.Much thanks again. Cool.
Thanks so much for the blog post. Really Cool.
My brother recommended I might like this web site. He was totally right.
This post truly made my day. You can not imagine just how much
time I had spent for this info! Thanks!
Enjoyed every bit of your blog post. Awesome.
Very neat blog post.Thanks Again. Really Great.
With havin so much written content do you ever run into any problems of plagorism
or copyright infringement? My website has a lot of completely unique content I’ve either created myself or outsourced
but it seems a lot of it is popping it up all over the web without my authorization. Do you know any ways
to help reduce content from being stolen?
I’d certainly appreciate it.
Look at my web page – http://winkler-martin.de/messages/61849.html
Please let me know if you’re looking for a author for your blog.
You have some really great posts and I think I would
be a good asset. If you ever want to take some of the load off,
I’d really like to write some material for your blog in exchange for a
link back to mine. Please send me an email if interested.
Thanks!
I am so grateful for your blog. Really Cool.
Very good blog.Thanks Again. Really Cool.
Im grateful for the blog. Want more.
Really enjoyed this blog article.Thanks Again. Much obliged.
Write more, thats all I have to say. Literally, it seems as though you relied on the video
to make your point. You clearly know what youre talking about, why waste your intelligence on just posting videos to your blog when you could be giving us something enlightening
to read?
Thanks for the blog article.Really thank you! Cool.
Hey! Do you know if they make any plugins to assist with SEO?
I’m trying to get my blog to rank for some targeted keywords but I’m
not seeing very good success. If you know of any please share.
Cheers!
I was curious if you ever thought of changing the structure of your blog?
Its very well written; I love what youve got to say. But maybe you
could a little more in the way of content so people could connect with it better.
Youve got an awful lot of text for only having one or
two images. Maybe you could space it out better?
Thanks for sharing, this is a fantastic article post.Thanks Again. Much obliged.
I think this is a real great blog.Really looking forward to read more. Want more.
Thank you for your blog article.Thanks Again. Want more.
If you are going for finest contents like myself, just visit this web
site all the time because it presents quality contents, thanks
A round of applause for your blog article.Much thanks again. Much obliged.
Very informative blog post.Really looking forward to read more. Keep writing.
I absolutely love your site.. Pleasant colors & theme. Did you make this site yourself? Please reply back as I’m attempting to create my own personal blog and would like to find out where you got this from or what the theme is named. Thanks.
I seriously love your site.. Great colors & theme. Did you build this site yourself? Please reply back as I’m looking to create my own personal website and would love to find out where you got this from or just what the theme is named. Appreciate it.
May I simply say what a comfort to discover an individual who really knows what they’re talking about on the net. You actually realize how to bring a problem to light and make it important. More and more people must read this and understand this side of your story. I was surprised that you are not more popular given that you definitely have the gift.
Aw, this was a really nice post. Taking a few minutes and actual effort to create a top notch article… but what can I say… I hesitate a lot and don’t seem to get nearly anything done.
Great article! That is the kind of information that
are supposed to be shared across the internet. Disgrace on Google for no longer positioning this submit upper!
Come on over and discuss with my website . Thank you =)
Pretty! This has been an extremely wonderful post. Many thanks for supplying this information.
I am not sure where you’re getting your information, but
great topic. I needs to spend some time learning much more or understanding more.
Thanks for great info I was looking for this info for my mission.
Today, I went to the beach front with my children. I found a sea shell and gave it to my 4 year old
daughter and said “You can hear the ocean if you put this to your ear.” She put
the shell to her ear and screamed. There was a hermit crab inside and it pinched her ear.
She never wants to go back! LoL I know this is completely off topic but I had to tell
someone!
This is a topic that is close to my heart… Best wishes!
Exactly where are your contact details though?
Hello my family member! I want to say that this article is awesome,
nice written and include almost all vital infos. I would like to peer more posts like this .
I like the helpful info you supply in your articles. I will bookmark your weblog and take
a look at once more right here frequently. I am rather certain I’ll be
told plenty of new stuff right right here! Good luck for the following!
After looking into a few of the blog articles on your web page, I seriously like your technique of blogging. I saved as a favorite it to my bookmark webpage list and will be checking back in the near future. Take a look at my website too and tell me how you feel.
Good post. I am going through some of these issues as well..
Good post. I learn something totally new and challenging on sites I stumbleupon on a daily basis. It will always be interesting to read through articles from other writers and use something from their websites.
I truly love your website.. Excellent colors & theme. Did you create this website yourself? Please reply back as I’m wanting to create my very own site and want to find out where you got this from or just what the theme is called. Thanks!
I really love your blog.. Excellent colors & theme. Did you make this web site yourself? Please reply back as I’m hoping to create my very own blog and would love to find out where you got this from or what the theme is named. Appreciate it!
I used to be able to find good information from your blog articles.
Aw, this was a really good post. Spending some time and actual effort to generate a top notch article… but what can I say… I put things off a lot and don’t seem to get anything done.
Right here is the perfect webpage for anyone who wishes to find out about this topic. You understand a whole lot its almost tough to argue with you (not that I personally would want to…HaHa). You definitely put a fresh spin on a topic that has been written about for years. Excellent stuff, just great.
There is certainly a lot to find out about this subject. I really like all of the points you made.
Everyone loves it whenever people get together and share views. Great blog, keep it up!
Saved as a favorite, I love your website!
Hello! I simply would like to offer you a big thumbs up for the excellent info you have right here on this post. I will be returning to your blog for more soon.
Good article! We will be linking to this great article on our website. Keep up the good writing.
You’ve made some really good points there. I looked on the web to learn more about the issue and found most individuals will go along with your views on this site.
When I originally commented I clicked the “Notify me when new comments are added” checkbox and now
each time a comment is added I get several e-mails with the same comment.
Is there any way you can remove me from that service? Many thanks!
I’ve been surfing online more than 2 hours today, yet I never found any interesting article
like yours. It’s pretty worth enough for me. In my opinion, if
all site owners and bloggers made good content as you did, the web will be
a lot more useful than ever before.
We are a group of volunteers and starting a new scheme in our community.
Your website offered us with helpful info to work on. You’ve performed
a formidable task and our entire neighborhood will likely be thankful to you.
Very good info. Lucky me I found your blog by chance (stumbleupon). I’ve saved it for later.
I’m amazed, I must say. Seldom do I encounter a blog that’s both educative and amusing, and without a doubt, you’ve hit the nail on the head. The issue is something which not enough folks are speaking intelligently about. I’m very happy I found this during my search for something relating to this.
Everything is very open with a very clear explanation of the issues. It was definitely informative. Your website is very useful. Many thanks for sharing!
Hello there! I just would like to offer you a huge thumbs up for the great info you have got right here on this post. I will be coming back to your web site for more soon.
Hello, I believe your web site might be having browser compatibility issues. Whenever I look at your blog in Safari, it looks fine however, if opening in Internet Explorer, it’s got some overlapping issues. I just wanted to provide you with a quick heads up! Other than that, fantastic site.
Having read this I believed it was extremely informative. I appreciate you taking the time and energy to put this information together. I once again find myself personally spending way too much time both reading and posting comments. But so what, it was still worth it!
Hello there, There’s no doubt that your website may be having internet browser compatibility issues. Whenever I look at your web site in Safari, it looks fine however, when opening in Internet Explorer, it’s got some overlapping issues. I just wanted to give you a quick heads up! Besides that, excellent site!
Very good post. I will be going through some of these issues as well..
Excellent blog you have here.. It’s difficult to find excellent writing like yours nowadays. I really appreciate people like you! Take care!!
Hi, I do believe this is a great web site. I stumbledupon it 😉 I will revisit once again since I saved as a favorite it. Money and freedom is the greatest way to change, may you be rich and continue to guide other people.
Good blog post. I certainly appreciate this website. Stick with it!
Your style is really unique compared to other folks I have read stuff from. Thank you for posting when you have the opportunity, Guess I will just book mark this site.
Can I just say what a relief to uncover someone who genuinely understands what they are talking about on the net. You certainly understand how to bring a problem to light and make it important. More and more people must read this and understand this side of your story. I was surprised that you’re not more popular given that you certainly possess the gift.
Can I simply say what a comfort to uncover a person that genuinely knows what they are discussing on the net. You certainly know how to bring an issue to light and make it important. A lot more people need to check this out and understand this side of your story. I was surprised that you aren’t more popular given that you definitely have the gift.
Spot on with this write-up, I absolutely believe that this web site needs a great deal more attention. I’ll probably be back again to read more, thanks for the information.
Greetings, There’s no doubt that your website might be having web browser compatibility issues. Whenever I look at your web site in Safari, it looks fine however, when opening in I.E., it’s got some overlapping issues. I just wanted to provide you with a quick heads up! Apart from that, great site.
I truly love your site.. Excellent colors & theme. Did you build this site yourself? Please reply back as I’m trying to create my own website and would like to know where you got this from or exactly what the theme is named. Appreciate it!
You’re so interesting! I do not suppose I’ve truly read something like that before. So wonderful to discover somebody with original thoughts on this topic. Seriously.. thank you for starting this up. This site is something that is needed on the web, someone with a little originality.
An impressive share! I have just forwarded this onto a friend who was conducting a little research on this. And he in fact ordered me dinner due to the fact that I stumbled upon it for him… lol. So allow me to reword this…. Thank YOU for the meal!! But yeah, thanks for spending time to talk about this matter here on your web page.
I really love your website.. Very nice colors & theme. Did you build this amazing site yourself? Please reply back as I’m planning to create my own personal site and would love to know where you got this from or exactly what the theme is named. Many thanks.
Can I simply say what a comfort to find someone that really knows what they are talking about over the internet. You definitely realize how to bring an issue to light and make it important. More and more people really need to check this out and understand this side of the story. I was surprised that you are not more popular since you surely possess the gift.
doktor kbb
dr güncel
dr kandulu
dr şaban
This website was… how do I say it? Relevant!!
Finally I have found something that helped me. Thanks a lot!
Way cool! Some very valid points! I appreciate you penning this article plus the rest of the site is extremely good.
Howdy! I could have sworn I’ve been to this site before but after browsing through many of the posts I realized it’s new to me. Regardless, I’m definitely pleased I came across it and I’ll be book-marking it and checking back often.
Saved as a favorite, I love your site.
This excellent website really has all of the information I needed about this subject and didn’t know who to ask.
I’m impressed, I have to admit. Rarely do I come across a blog that’s both educative and amusing, and let me tell you, you have hit the nail on the head. The problem is something which not enough men and women are speaking intelligently about. I’m very happy that I found this in my search for something concerning this.
smile
esteworld
Good article! We are linking to this particularly great content on our website. Keep up the great writing.
Having read this I believed it was really informative. I appreciate you spending some time and energy to put this information together. I once again find myself personally spending a lot of time both reading and commenting. But so what, it was still worthwhile!
medhair
I’m amazed, I have to admit. Rarely do I encounter a blog that’s both equally educative and amusing, and without a doubt, you’ve hit the nail on the head. The problem is something that not enough men and women are speaking intelligently about. I am very happy that I stumbled across this during my search for something regarding this.
May I simply say what a comfort to discover somebody that actually knows what they’re discussing on the internet. You definitely know how to bring an issue to light and make it important. More people really need to read this and understand this side of the story. I was surprised you aren’t more popular since you definitely have the gift.
sapphire hair clinic
I think that everything published was actually very logical.
But, what about this? suppose you wrote a catchier post title?
I mean, I don’t want to tell you how to run your blog, but suppose you added
a title to possibly get a person’s attention? I mean 構造設備のCSV実施方法 | お役立ち情報 is a little boring.
You ought to glance at Yahoo’s front page and note how they
write article titles to get viewers interested.
You might add a related video or a pic or two to grab people interested
about what you’ve got to say. In my opinion, it might make your posts a
little livelier.
An outstanding share! I have just forwarded this onto a coworker who was conducting a little research on this. And he in fact ordered me breakfast because I stumbled upon it for him… lol. So allow me to reword this…. Thank YOU for the meal!! But yeah, thanx for spending the time to discuss this matter here on your site.
Quality articles is the main to interest the people to pay a visit the web site,
that’s what this site is providing.
Hi Dear, are you in fact visiting this web site on a regular basis, if so then you
will without doubt get good knowledge.
Nice post. I learn something totally new and challenging on blogs I stumbleupon everyday. It’s always useful to read through articles from other writers and use something from other sites.
Everyone loves it when folks come together and share ideas. Great blog, continue the good work!
Link exchange is nothing else but it is simply placing the
other person’s webpage link on your page at appropriate place and other person will also do similar for you.
Hi there! Someone in my Myspace group shared this site with us so I came
to give it a look. I’m definitely loving the information. I’m book-marking and will be tweeting
this to my followers! Terrific blog and wonderful design.
Can I simply just say what a relief to uncover a person that truly understands what they are discussing online. You definitely realize how to bring a problem to light and make it important. More and more people should check this out and understand this side of your story. I was surprised you’re not more popular because you certainly possess the gift.
Hi there! This blog post could not be written any better! Looking at this article reminds me of my previous roommate! He always kept talking about this. I most certainly will forward this article to him. Pretty sure he’s going to have a great read. I appreciate you for sharing!
Saved as a favorite, I really like your blog.
Aw, this was an exceptionally nice post. Taking a few minutes and actual effort to make a superb article… but what can I say… I procrastinate a lot and don’t manage to get anything done.
Greetings! Very helpful advice within this article! It is the little changes that produce the most important changes. Thanks a lot for sharing!
This is a great tip particularly to those fresh to the blogosphere. Brief but very precise information… Thank you for sharing this one. A must read article!
Hello there! This post could not be written any better! Looking at this article reminds me of my previous roommate! He constantly kept preaching about this. I am going to forward this article to him. Fairly certain he’s going to have a great read. I appreciate you for sharing!
Hello everyone, it’s my first pay a quick visit at this website,
and piece of writing is in fact fruitful in support of me, keep up posting these types of articles.
Hey there! I’ve been reading your blog for a long time now and finally got the bravery
to go ahead and give you a shout out from Humble Texas!
Just wanted to say keep up the excellent work!
What’s up everybody, here every person is sharing these kinds of familiarity, therefore it’s fastidious to read this webpage,
and I used to pay a quick visit this webpage every day.
Keep on working, great job!
Nice post. I learn something new and challenging on sites I stumbleupon everyday. It’s always exciting to read articles from other writers and use something from other sites.
You’ve made some decent points there. I checked on the web for more information about the issue and found most people will go along with your views on this site.
Very good information. Lucky me I came across your website by chance (stumbleupon). I’ve saved as a favorite for later.
I love looking through an article that will make men and women think. Also, thank you for allowing me to comment.
I like looking through a post that can make people think. Also, many thanks for permitting me to comment.
Magnificent beat ! I wish to apprentice at the same time
as you amend your website, how can i subscribe for a blog site?
The account aided me a acceptable deal. I have been tiny bit acquainted of this your broadcast offered
vibrant transparent idea
This is a very good tip especially to those new to the blogosphere. Short but very precise information… Many thanks for sharing this one. A must read article.
Hi! I could have sworn I’ve visited this website before but after browsing through some of the articles I realized it’s new to me. Regardless, I’m certainly delighted I found it and I’ll be bookmarking it and checking back often!
After checking out a handful of the blog articles on your site, I truly appreciate your way of writing a blog. I added it to my bookmark site list and will be checking back soon. Take a look at my website too and tell me how you feel.
This site was… how do I say it? Relevant!! Finally I have found something which helped me. Kudos!
Having read this I thought it was really informative. I appreciate you finding the time and energy to put this content together. I once again find myself spending a significant amount of time both reading and leaving comments. But so what, it was still worth it!
bookmarked!!, I really like your website.
It’s awesome designed for me to have a web site, which is useful designed
for my know-how. thanks admin
I blog frequently and I seriously appreciate your content. The article has really peaked my interest. I’m going to take a note of your website and keep checking for new details about once a week. I opted in for your Feed as well.
Howdy! I could have sworn I’ve been to your blog before but after looking at many of the articles I realized it’s new to me. Regardless, I’m certainly pleased I stumbled upon it and I’ll be bookmarking it and checking back regularly!
That is a very good tip particularly to those new to the blogosphere. Simple but very precise information… Appreciate your sharing this one. A must read post!
There is certainly a great deal to know about this issue. I like all of the points you have made.
Very nice blog post. I absolutely love this site. Keep writing!
Hi there! I just wish to offer you a huge thumbs up for the great info you’ve got here on this post. I am coming back to your site for more soon.
Good day! I could have sworn I’ve been to this blog before but after looking at a few of the posts I realized it’s new to me. Anyways, I’m certainly delighted I stumbled upon it and I’ll be bookmarking it and checking back regularly.
I got this site from my pal who told me regarding this web
page and at the moment this time I am browsing this website and reading very informative articles here.
Keep this going please, great job! vpn https://www.highlandguides.com
Everyone loves what you guys tend to be up too.
Such clever work and coverage! Keep up the amazing works
guys I’ve included you guys to my own blogroll.
I love it when people get together and share thoughts. Great website, continue the good work!
Excellent article! We are linking to this particularly great article on our site. Keep up the great writing.
This site was… how do you say it? Relevant!! Finally I have found something that helped me. Thanks.
I like it when individuals come together and share opinions. Great blog, stick with it!
Hierüber lässt sich auch der 400% Einzahlungsbonus bis zu 80€ mit demBetano Bonus Code„NTVSLOT“ unkompliziert einlösen. Bereits ab einer Einzahlung von 10€ kann der Wildz Einzahlungsbonus aktiviert werden. Dabei kann der Tipico Willkommensbonus mit sämtlichen Zahlungsoptionen und einer Einzahlung von mindestens 10€ aktiviert werden. Dies zeigt sich auch in der Kooperation mit sicheren und etablierten Zahlungsdienstleistern. Neue Spieler können sich somit einen 100% Bonus bis zu 200€ sowie zusätzlich 50 Freispiele für denBook of DeadSlot sichern.
Zahlung über myPaysafe bestätigen und schon ist das Geld auf dem Spielkonto. Bereits ab 5 Euro ist man dabei und qualifiziert sich zudem für das Willkommenspaket mit bis zu 100 Freispielen. Einzahlungen ab 10 € sind sofort auf eurem Konto und der Willkommensbonus von bis zu 100 € plus 10 Freispiele täglich wartet ebenfalls auf euch, natürlich mit fairen Umsatzbedingungen. PaysafeCard bietet euch die Freiheit, ohne die Angabe von Bankdaten oder Kreditkarteninfos direkt ins Spiel zu starten. Für sicheres Spielen sorgt unter anderem die Lizenz der GGL. Die Mindesteinzahlung liegt bei 1€, sodass auch diejenigen mit besonders kleinem Budget am Spielspaß teilnehmen können.
References:
https://online-spielhallen.de/playfina-casino-erfahrungen-ein-detaillierter-bericht/
Good post. I learn something new and challenging on sites I stumbleupon everyday. It’s always useful to read through articles from other writers and use something from other websites.
Aw, this was a really good post. Taking a few minutes and actual effort to make a good article… but what can I say… I put things off a lot and don’t manage to get anything done.
You ought to be a part of a contest for one of the most useful sites on the internet. I will highly recommend this website!
Howdy, I do think your web site could possibly be having browser compatibility issues. Whenever I look at your site in Safari, it looks fine but when opening in I.E., it’s got some overlapping issues. I simply wanted to provide you with a quick heads up! Aside from that, great website!
An impressive share! I’ve just forwarded this onto a coworker who was conducting a little research on this. And he in fact bought me dinner due to the fact that I discovered it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanx for spending some time to talk about this issue here on your site.
I’m pretty pleased to uncover this site. I need to to thank you for ones time just for this wonderful read!! I definitely enjoyed every bit of it and I have you book-marked to look at new stuff on your web site.
Great post! We are linking to this particularly great post on our site. Keep up the great writing.
It’s difficult to find knowledgeable people in this particular subject, but you seem like you know what you’re talking about! Thanks
Your style is unique in comparison to other folks I have read stuff from. Thank you for posting when you’ve got the opportunity, Guess I will just book mark this page.
Your style is very unique compared to other people I’ve read stuff from. Many thanks for posting when you’ve got the opportunity, Guess I will just bookmark this web site.
Good day! I just wish to offer you a huge thumbs up for your great information you’ve got here on this post. I will be returning to your blog for more soon.
Hello everyone, it’s my first pay a quick visit at this
site, and paragraph is genuinely fruitful designed for me, keep up
posting these content.
Pretty! This has been an extremely wonderful article. Thanks for providing these details.
What’s up, this weekend is good for me, for the reason that this moment i am reading this great informative paragraph
here at my residence.
After I initially commented I seem to have clicked on the -Notify me when new comments are added- checkbox and from now on each time a comment is added I receive 4 emails with the exact same comment. There has to be an easy method you can remove me from that service? Thanks.
Hello there, I believe your web site could possibly be having web browser compatibility issues. When I take a look at your web site in Safari, it looks fine however, if opening in Internet Explorer, it’s got some overlapping issues. I simply wanted to give you a quick heads up! Other than that, excellent blog!
Great information. Lucky me I discovered your site by accident (stumbleupon). I have saved as a favorite for later!
Howdy! I could have sworn I’ve been to this blog before but after going through some of the posts I realized it’s new to me. Nonetheless, I’m certainly happy I stumbled upon it and I’ll be bookmarking it and checking back frequently.
Very good information. Lucky me I discovered your blog by chance (stumbleupon). I have saved it for later!
Hi, I do think this is an excellent blog. I stumbledupon it 😉 I will return once again since I book-marked it. Money and freedom is the best way to change, may you be rich and continue to help other people.
A great place for an evening drink, at the bar closest to the atmosphere of the gaming area. Bolters Grill is part of the same precinct beside The Villas accommodation that includes Bolters Pizzeria, Bar and Bottle Shop. Whether you’re considering an end-of-year or mid-year gathering, a big announcement, or an anniversary or birthday celebration, book ahead for breakfast, lunch or dinner at Links today.
Our 18-hole mini-golf course will also be a big hit with adults and families alike. Includes putter and golf ball hire. Why not try a round of mini golf at The Range? Range Bay pricing is for 1 – 6 people and includes club hireWhen are peak and non-peak times? Select your preferred time, with options across peak, and non-peak periods.
Just visit Member Services at Country Club Tasmania or Wrest Point Hotel & Casino today with a valid photo ID to be signed up. With exclusive access to an increased range of special benefits. Enjoy discounted accommodation at Wrest Point and Country Club Tasmania. You can even browse our Member Showcase for vouchers and all kinds of items including lifestyle vouchers and household products – even Christmas hampers!
References:
https://blackcoin.co/play-99-casino-in-australia-real-money-gaming-experience/
Good day! I could have sworn I’ve visited your blog before but after browsing through many of the articles I realized it’s new to me. Nonetheless, I’m definitely happy I came across it and I’ll be bookmarking it and checking back frequently!
Oh my goodness! Impressive article dude! Many thanks, However I am having problems with your RSS. I don’t understand the reason why I am unable to join it. Is there anyone else having identical RSS issues? Anyone that knows the answer will you kindly respond? Thanks.
Membership to the Crown Rewards program is free, offering access to gaming areas, especially for Black and Platinum members. The staff are also helpful and readily available to guide players to accessible areas. So, you can conveniently enjoy the games without worrying about carrying physical cash. The Crown Sydney Casino dealers are knowledgeable about the games.
Crown Rewards members who wish to visit Casino Crown Sydney must have a Crown Sydney Casino membership. Please note that Crown Rewards Members wishing to visit Crown Sydney Casino must have a Casino Sydney Crown membership. This unique reward system offers you endless ways to earn and enjoy the benefits you love. The gameplay is conducted on an individual virtual terminal. The game requires basic skills and a mathematical mindset.
References:
https://blackcoin.co/are-bitcoin-casinos-better-than-normal-casinos/
After looking at a handful of the blog posts on your blog, I truly like your technique of blogging. I saved it to my bookmark website list and will be checking back soon. Take a look at my web site too and let me know what you think.
Excellent post. I am going through a few of these issues as well..
Very good info. Lucky me I ran across your website by chance (stumbleupon). I have bookmarked it for later.
Can I simply say what a relief to discover somebody that truly understands what they are talking about on the web. You definitely understand how to bring a problem to light and make it important. More and more people must read this and understand this side of the story. I was surprised that you are not more popular given that you certainly possess the gift.
Hi! I could have sworn I’ve visited your blog before but after looking at many of the articles I realized it’s new to me. Nonetheless, I’m definitely pleased I discovered it and I’ll be book-marking it and checking back frequently!
I want to to thank you for this very good read!! I definitely enjoyed every bit of it. I have got you book-marked to look at new things you post…
Having read this I thought it was rather enlightening. I appreciate you spending some time and effort to put this article together. I once again find myself personally spending a lot of time both reading and leaving comments. But so what, it was still worth it!
Hi, I do think this is a great website. I stumbledupon it 😉 I’m going to return yet again since I saved as a favorite it. Money and freedom is the greatest way to change, may you be rich and continue to help others.
I was able to find good information from your blog articles.
Greetings! Very helpful advice within this post! It is the little changes that make the most important changes. Thanks a lot for sharing!
Good blog you’ve got here.. It’s hard to find excellent writing like yours nowadays. I really appreciate people like you! Take care!!
Excellent site you have here.. It’s difficult to find high-quality writing like yours nowadays. I seriously appreciate people like you! Take care!!
I really love your website.. Great colors & theme. Did you make this amazing site yourself? Please reply back as I’m planning to create my very own site and would like to find out where you got this from or exactly what the theme is called. Kudos.
us online casinos that accept paypal
References:
https://www.madeinna.org/profile/amiemacdevitt1
paypal online casinos
References:
https://recruitment.econet.co.zw/employer/paypal-casinos-2025-best-online-casinos-accepting-paypal/
After I originally commented I appear to have clicked on the -Notify me when new comments are added- checkbox and from now on whenever a comment is added I recieve four emails with the same comment. There has to be an easy method you can remove me from that service? Kudos.
online real casino paypal
References:
https://classifylistings.com/index.php?page=user&action=pub_profile&id=309278&item_type=active&per_page=16
us online casinos paypal
References:
https://precisionscans.net/employer/top-10-games-in-online-casinos/
Very quickly this web page will be famous amid all blogging users, due to it’s
good articles
Very good blog post. I certainly appreciate this website. Stick with it!
Hello! I could have sworn I’ve visited this website before but after browsing through some of the articles I realized it’s new to me. Nonetheless, I’m definitely pleased I came across it and I’ll be bookmarking it and checking back often!
This website really has all of the information I wanted concerning this subject and didn’t know who to ask.
Can I simply just say what a comfort to find someone who truly understands what they are discussing on the internet. You definitely know how to bring an issue to light and make it important. A lot more people really need to look at this and understand this side of the story. I can’t believe you’re not more popular because you definitely possess the gift.
You are so awesome! I don’t think I’ve truly read something like this before. So great to discover somebody with original thoughts on this subject matter. Really.. thank you for starting this up. This website is one thing that is needed on the web, someone with some originality.
Greetings! Very helpful advice within this article! It’s the little changes that will make the most important changes. Thanks a lot for sharing!
Having read this I believed it was rather informative. I appreciate you spending some time and effort to put this content together. I once again find myself spending way too much time both reading and leaving comments. But so what, it was still worthwhile.
After going over a number of the articles on your web site, I really appreciate your way of writing a blog. I book marked it to my bookmark website list and will be checking back in the near future. Please visit my website as well and let me know your opinion.
You’re so awesome! I don’t suppose I have read anything like this before. So nice to discover somebody with a few genuine thoughts on this subject matter. Really.. thank you for starting this up. This site is something that’s needed on the web, someone with a little originality.
This web site truly has all of the information I wanted concerning this subject and didn’t know who to ask.
This is a really good tip particularly to those fresh to the blogosphere. Short but very precise information… Thanks for sharing this one. A must read article.
I’m more than happy to find this site. I need to to thank you for ones time for this wonderful read!! I definitely appreciated every bit of it and i also have you saved as a favorite to see new things on your website.
When I originally left a comment I appear to have clicked the -Notify me when new comments are added- checkbox and now each time a comment is added I get 4 emails with the same comment. There has to be a means you are able to remove me from that service? Thank you.
I’m excited to uncover this great site. I want to to thank you for ones time just for this fantastic read!! I definitely liked every little bit of it and i also have you saved to fav to see new information in your site.
You ought to be a part of a contest for one of the finest websites on the net. I’m going to recommend this blog!
A motivating discussion is definitely worth comment. I believe that you should write more on this subject matter, it may not be a taboo subject but typically people do not discuss such issues. To the next! Kind regards!
After checking out a handful of the articles on your web site, I seriously like your way of blogging. I bookmarked it to my bookmark website list and will be checking back in the near future. Please visit my website as well and let me know what you think.
Hello there! This blog post could not be written any better! Looking through this article reminds me of my previous roommate! He constantly kept preaching about this. I’ll forward this information to him. Pretty sure he will have a good read. Many thanks for sharing!
An outstanding share! I have just forwarded this onto a coworker who has been conducting a little research on this. And he actually ordered me lunch simply because I found it for him… lol. So allow me to reword this…. Thanks for the meal!! But yeah, thanks for spending some time to talk about this issue here on your internet site.
After I initially commented I appear to have clicked on the -Notify me when new comments are added- checkbox and now whenever a comment is added I recieve 4 emails with the exact same comment. Is there a means you can remove me from that service? Thanks a lot.
This is a topic that’s near to my heart… Best wishes! Exactly where can I find the contact details for questions?
Excellent article. I absolutely love this site. Continue the good work!
I truly love your website.. Pleasant colors & theme. Did you develop this website yourself? Please reply back as I’m attempting to create my own personal site and would love to learn where you got this from or exactly what the theme is called. Kudos.
This is a topic that is close to my heart… Thank you! Where can I find the contact details for questions?
After checking out a handful of the articles on your website, I honestly like your way of blogging. I added it to my bookmark site list and will be checking back soon. Please check out my web site too and let me know what you think.
It’s in fact very complex in this busy life to listen news on Television, therefore
I only use web for that purpose, and obtain the newest news.
Very good information. Lucky me I found your website by chance (stumbleupon). I’ve saved it for later.
Way cool! Some very valid points! I appreciate you writing this write-up and also the rest of the website is also very good.
A motivating discussion is worth comment. I believe that you ought to write more on this subject matter, it may not be a taboo matter but usually folks don’t discuss such subjects. To the next! Cheers!
Greetings! Very helpful advice in this particular article! It is the little changes which will make the most important changes. Many thanks for sharing!
I’m pretty pleased to discover this page. I wanted to thank you for ones time just for this wonderful read!! I definitely savored every bit of it and I have you book-marked to check out new information in your blog.
Next time I read a blog, Hopefully it doesn’t disappoint me as much as this one. I mean, Yes, it was my choice to read, nonetheless I truly thought you would probably have something useful to talk about. All I hear is a bunch of complaining about something that you can fix if you weren’t too busy looking for attention.
I really like it when individuals get together and share thoughts. Great blog, continue the good work!
This is a topic that’s close to my heart… Take care! Where are your contact details though?
Way cool! Some extremely valid points! I appreciate you penning this article and the rest of the site is very good.
Click Here – I didn’t expect to find such helpful content. Thanks a lot!
An outstanding share! I have just forwarded this onto a co-worker who has been conducting a little research on this. And he in fact ordered me dinner due to the fact that I stumbled upon it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanks for spending time to talk about this topic here on your site.
Very nice write-up. I definitely appreciate this website. Stick with it!
Very good info. Lucky me I found your website by accident (stumbleupon). I have saved as a favorite for later.
Hello there! I just want to offer you a huge thumbs up for the great information you’ve got here on this post. I am coming back to your web site for more soon.
Hi there! I could have sworn I’ve been to this site before but after going through a few of the articles I realized it’s new to me. Regardless, I’m certainly pleased I found it and I’ll be book-marking it and checking back regularly!
Hi, I do think this is a great website. I stumbledupon it 😉 I’m going to come back once again since i have book marked it. Money and freedom is the greatest way to change, may you be rich and continue to help others.
An outstanding share! I’ve just forwarded this onto a coworker who has been doing a little homework on this. And he actually ordered me lunch simply because I found it for him… lol. So let me reword this…. Thank YOU for the meal!! But yeah, thanx for spending the time to discuss this matter here on your website.
Greetings! Very useful advice within this article! It is the little changes that will make the biggest changes. Thanks for sharing!
Next time I read a blog, I hope that it does not fail me as much as this one. After all, Yes, it was my choice to read, but I really thought you would have something interesting to talk about. All I hear is a bunch of whining about something that you could fix if you weren’t too busy seeking attention.
Can I simply say what a comfort to uncover an individual who actually knows what they’re discussing on the web. You certainly understand how to bring a problem to light and make it important. More and more people have to check this out and understand this side of your story. I was surprised that you aren’t more popular given that you surely have the gift.
This is a topic that is near to my heart… Cheers! Exactly where are your contact details though?
Spot on with this write-up, I truly believe this amazing site needs a lot more attention. I’ll probably be returning to read through more, thanks for the advice.
An outstanding share! I’ve just forwarded this onto a coworker who had been doing a little research on this. And he in fact ordered me dinner due to the fact that I discovered it for him… lol. So allow me to reword this…. Thanks for the meal!! But yeah, thanks for spending time to talk about this matter here on your website.
What’s up to all, because I am truly eager of reading this
website’s post to be updated on a regular basis. It carries pleasant information.
Pretty! This has been an incredibly wonderful article. Thank you for providing this info.
There is definately a great deal to learn about this topic. I really like all the points you have made.
Hi there! This article could not be written much better! Looking through this article reminds me of my previous roommate! He constantly kept preaching about this. I am going to send this information to him. Pretty sure he will have a good read. Thanks for sharing!
Great article! We are linking to this great article on our website. Keep up the great writing.
Oh my goodness! Awesome article dude! Thank you, However I am having difficulties with your RSS. I don’t know why I am unable to subscribe to it. Is there anyone else getting identical RSS issues? Anybody who knows the answer can you kindly respond? Thanks!!
It’s hard to come by well-informed people about this subject, however, you seem like you know what you’re talking about! Thanks
Your style is very unique compared to other people I have read stuff from. Thanks for posting when you have the opportunity, Guess I’ll just bookmark this blog.
Hello there! I could have sworn I’ve been to this web site before but after going through many of the articles I realized it’s new to me. Anyhow, I’m certainly happy I stumbled upon it and I’ll be bookmarking it and checking back regularly!
Saved as a favorite, I really like your blog.
Howdy, I believe your site could possibly be having web browser compatibility problems. When I look at your blog in Safari, it looks fine but when opening in I.E., it’s got some overlapping issues. I merely wanted to provide you with a quick heads up! Other than that, great site!
Howdy! I just want to offer you a big thumbs up for your excellent info you’ve got right here on this post. I am returning to your blog for more soon.
Hi! This is my 1st comment here so I just wanted to give a quick shout out and say
I truly enjoy reading through your posts. Can you suggest any other blogs/websites/forums that go over the same
topics? Appreciate it!
Good day! I simply would like to give you a huge thumbs up for the excellent info you have here on this post. I will be coming back to your website for more soon.
Hi there! Someone in my Myspace group shared this site with us
so I came to give it a look. I’m definitely enjoying the information. I’m bookmarking and will be tweeting this to my
followers! Terrific blog and superb design and style.
Hey there! I simply wish to offer you a big thumbs up for the great info you have right here on this post. I’ll be returning to your web site for more soon.
I like reading a post that will make people think. Also, thank you for permitting me to comment.
I quite like reading through a post that can make people think. Also, thank you for allowing for me to comment.
I really love your website.. Pleasant colors & theme. Did you develop this amazing site yourself? Please reply back as I’m trying to create my own personal site and want to learn where you got this from or what the theme is named. Thanks.
I like reading an article that can make men and women think. Also, many thanks for permitting me to comment.
That is a great tip particularly to those fresh to the blogosphere. Simple but very accurate info… Thank you for sharing this one. A must read post.
Hello there! This blog post could not be written any better! Reading through this article reminds me of my previous roommate! He always kept talking about this. I’ll forward this article to him. Fairly certain he’s going to have a good read. I appreciate you for sharing!
Great site you have here.. It’s difficult to find quality writing like yours nowadays. I really appreciate people like you! Take care!!
When I originally commented I appear to have clicked the -Notify me when new comments are added- checkbox and now each time a comment is added I receive four emails with the exact same comment. There has to be a means you are able to remove me from that service? Many thanks.
This is a topic that’s close to my heart… Many thanks! Exactly where are your contact details though?
Can I just say what a relief to discover somebody who genuinely knows what they’re talking about online. You certainly understand how to bring an issue to light and make it important. More and more people ought to look at this and understand this side of your story. I can’t believe you aren’t more popular because you certainly possess the gift.
I’m very happy to uncover this site. I wanted to thank you for your time for this wonderful read!! I definitely loved every little bit of it and I have you bookmarked to see new things on your website.
Hi, I think your site might be having web browser compatibility problems. Whenever I look at your site in Safari, it looks fine however, if opening in Internet Explorer, it has some overlapping issues. I merely wanted to provide you with a quick heads up! Other than that, great website.
Good blog you have got here.. It’s difficult to find high-quality writing like yours these days. I honestly appreciate people like you! Take care!!
Oh my goodness! Incredible article dude! Many thanks, However I am experiencing problems with your RSS. I don’t know the reason why I can’t subscribe to it. Is there anybody having similar RSS issues? Anybody who knows the answer can you kindly respond? Thanks!
You’ve made some good points there. I looked on the internet to learn more about the issue and found most people will go along with your views on this website.
Your style is very unique compared to other people I’ve read stuff from. Many thanks for posting when you’ve got the opportunity, Guess I’ll just book mark this site.
May I simply say what a comfort to find somebody that truly understands what they’re talking about on the internet. You actually realize how to bring an issue to light and make it important. More and more people should check this out and understand this side of your story. It’s surprising you are not more popular since you most certainly have the gift.
This site certainly has all of the information I needed concerning this subject and didn’t know who to ask.
I really love your website.. Excellent colors & theme. Did you make this website yourself? Please reply back as I’m looking to create my own personal blog and would love to know where you got this from or just what the theme is named. Many thanks.
I need to to thank you for this great read!! I certainly loved every bit of it. I have got you book-marked to look at new stuff you post…
Pretty! This has been a really wonderful article. Thank you for supplying this info.
I need to to thank you for this great read!! I absolutely enjoyed every little bit of it. I’ve got you book marked to look at new stuff you post…
You need to take part in a contest for one of the finest sites on the web. I am going to highly recommend this web site!
Follow my blog – I didn’t expect to find such helpful content. Thanks a lot!
This is a very good tip especially to those fresh to the blogosphere. Brief but very accurate information… Many thanks for sharing this one. A must read post!
Good post. I learn something totally new and challenging on websites I stumbleupon everyday. It’s always interesting to read through content from other writers and use something from other websites.
This site was… how do you say it? Relevant!! Finally I’ve found something which helped me. Kudos!
Having read this I thought it was really enlightening. I appreciate you finding the time and energy to put this content together. I once again find myself personally spending way too much time both reading and posting comments. But so what, it was still worthwhile!
This is a topic that’s close to my heart… Cheers! Exactly where can I find the contact details for questions?
The very next time I read a blog, I hope that it won’t disappoint me as much as this particular one. I mean, I know it was my choice to read through, but I genuinely thought you’d have something useful to say. All I hear is a bunch of whining about something that you can fix if you were not too busy searching for attention.
Your style is really unique compared to other folks I’ve read stuff from. I appreciate you for posting when you have the opportunity, Guess I’ll just book mark this site.
There’s definately a lot to know about this topic. I love all of the points you made.
Right here is the perfect web site for everyone who really wants to understand this topic. You realize a whole lot its almost hard to argue with you (not that I personally will need to…HaHa). You certainly put a brand new spin on a subject that’s been discussed for a long time. Great stuff, just wonderful.
Hi, I do think this is an excellent site. I stumbledupon it 😉 I am going to return yet again since I book marked it. Money and freedom is the best way to change, may you be rich and continue to help other people.
That is a really good tip especially to those fresh to the blogosphere. Short but very accurate information… Thank you for sharing this one. A must read article!
Hello there, I do believe your website could possibly be having browser compatibility problems. When I look at your website in Safari, it looks fine but when opening in I.E., it’s got some overlapping issues. I simply wanted to provide you with a quick heads up! Apart from that, great site!
I appreciate you sharing this article.Really looking forward to read more. Awesome.
Excellent blog post. I absolutely appreciate this site. Thanks!
Hi there! I could have sworn I’ve been to this website before but after looking at many of the articles I realized it’s new to me. Nonetheless, I’m certainly happy I stumbled upon it and I’ll be book-marking it and checking back often.
Hi, I do think this is an excellent website. I stumbledupon it 😉 I will revisit once again since I saved as a favorite it. Money and freedom is the greatest way to change, may you be rich and continue to help others.
bookmarked!!, I love your website!
I’m impressed, I have to admit. Rarely do I come across a blog that’s equally educative and interesting, and let me tell you, you have hit the nail on the head. The issue is something that not enough folks are speaking intelligently about. I am very happy I came across this during my hunt for something relating to this.
Explore the mechanics behind Gamified Mining and see how participation can become a repeatable habit. Understand why engagement-driven distribution can build stronger narratives than hype cycles.
I absolutely love your site.. Very nice colors & theme. Did you make this web site yourself? Please reply back as I’m attempting to create my very own blog and would like to know where you got this from or exactly what the theme is called. Kudos.
Hello there! I could have sworn I’ve visited this web site before but after going through some of the posts I realized it’s new to me. Nonetheless, I’m definitely delighted I found it and I’ll be bookmarking it and checking back often.
I could not resist commenting. Well written!
There is certainly a lot to find out about this topic. I love all of the points you’ve made.
Hi there, I think your website may be having browser compatibility problems. When I take a look at your website in Safari, it looks fine however, if opening in IE, it’s got some overlapping issues. I just wanted to provide you with a quick heads up! Other than that, excellent blog!
The very next time I read a blog, I hope that it doesn’t fail me just as much as this particular one. I mean, Yes, it was my choice to read through, but I really believed you would probably have something useful to say. All I hear is a bunch of complaining about something that you can fix if you weren’t too busy searching for attention.
I like looking through a post that will make men and women think. Also, thank you for allowing for me to comment.
I’m very happy to discover this website. I need to to thank you for ones time for this wonderful read!! I definitely loved every bit of it and I have you saved to fav to see new information on your blog.
It’s nearly impossible to find knowledgeable people on this topic, however, you sound like you know what you’re talking about! Thanks
Hi, I do think this is an excellent blog. I stumbledupon it 😉 I may return once again since I saved as a favorite it. Money and freedom is the best way to change, may you be rich and continue to guide other people.
Having read this I thought it was rather enlightening. I appreciate you taking the time and effort to put this short article together. I once again find myself spending a lot of time both reading and posting comments. But so what, it was still worthwhile.
Your style is really unique compared to other people I’ve read stuff from. Thank you for posting when you’ve got the opportunity, Guess I will just bookmark this web site.
I’m amazed, I have to admit. Seldom do I encounter a blog that’s both educative and interesting, and let me tell you, you have hit the nail on the head. The problem is something not enough men and women are speaking intelligently about. Now i’m very happy I came across this during my hunt for something relating to this.
Having read this I thought it was rather enlightening. I appreciate you finding the time and energy to put this article together. I once again find myself spending way too much time both reading and commenting. But so what, it was still worth it!
I really like it when folks get together and share ideas. Great website, keep it up!
A motivating discussion is worth comment. I do believe that you should publish more on this topic, it might not be a taboo subject but usually folks don’t speak about such issues. To the next! Cheers!
Hello there! This blog post could not be written much better! Going through this article reminds me of my previous roommate! He constantly kept preaching about this. I’ll send this information to him. Pretty sure he’s going to have a great read. Many thanks for sharing!
Hi, I do believe this is a great web site. I stumbledupon it 😉 I am going to return yet again since i have bookmarked it. Money and freedom is the greatest way to change, may you be rich and continue to guide other people.
Good post. I learn something totally new and challenging on blogs I stumbleupon everyday. It will always be helpful to read through articles from other writers and practice something from their sites.
Dive into BNB Pump.fun for a practical look at how token creators are leveraging BNB Chain’s low fees and fast transactions.
I need to to thank you for this good read!! I certainly loved every little bit of it. I’ve got you bookmarked to look at new stuff you post…
There’s certainly a lot to learn about this issue. I like all the points you made.
I couldn’t refrain from commenting. Well written!
I was able to find good information from your blog articles.
Nice post. I learn something new and challenging on blogs I stumbleupon everyday. It’s always interesting to read content from other authors and practice something from other sites.
This blog was… how do you say it? Relevant!! Finally I’ve found something which helped me. Thanks a lot.
A motivating discussion is worth comment. I believe that you need to write more on this subject matter, it may not be a taboo matter but usually people don’t discuss such issues. To the next! All the best.
I truly love your site.. Great colors & theme. Did you develop this web site yourself? Please reply back as I’m hoping to create my very own site and want to learn where you got this from or what the theme is named. Appreciate it.
You’ve made some good points there. I checked on the internet to learn more about the issue and found most individuals will go along with your views on this web site.
Fine way of telling, and pleasant paragraph to take data regarding my presentation subject
matter, which i am going to present in school.
I was able to find good info from your blog posts.
Everything is very open with a very clear explanation of the challenges. It was definitely informative. Your website is very useful. Thank you for sharing!
Next time I read a blog, Hopefully it doesn’t fail me as much as this one. I mean, Yes, it was my choice to read through, however I actually thought you would have something useful to say. All I hear is a bunch of moaning about something you could possibly fix if you weren’t too busy looking for attention.
Excellent blog you’ve got here.. It’s hard to find quality writing like yours nowadays. I really appreciate individuals like you! Take care!!
This is a topic that is close to my heart… Many thanks! Where can I find the contact details for questions?